PERSONAL Sign in with your SPIE account to access your personal subscriptions or to use specific features such as save to my library, sign up for alerts, save searches, etc.
Pulsed infrared (IR) laser energy has been shown to modulate neurological activity through both stimulation and inhibition of action potentials. While the mechanism(s) behind this phenomenon is (are) not completely understood, certain hypotheses suggest that the rise in temperature from IR exposure could activate temperature- or pressure-sensitive ion channels or create pores in the cellular outer membrane, allowing an influx of typically plasma-membrane-impermeant ions. Studies using fluorescent intensity-based calcium ion (Ca2+) sensitive dyes show changes in Ca2+ levels after various IR stimulation parameters, which suggests that Ca2+ may originate from the external solution. However, activation of intracellular signaling pathways has also been demonstrated, indicating a more complex mechanism of increasing intracellular Ca2+ concentration. We quantified the Ca2+ mobilization in terms of influx from the external solution and efflux from intracellular organelles using Fura-2 and a high-speed ratiometric imaging system that rapidly alternates the dye excitation wavelengths. Using nonexcitable Chinese hamster ovarian (CHO-hM1) cells and neuroblastoma-glioma (NG108) cells, we demonstrate that intracellular IP3 receptors play an important role in the IR-induced Ca2+, with the
1.
Introduction
Application of infrared (IR) laser pulses with wavelengths ranging from 1.4 to and pulse durations in the order of micro- to milliseconds has been shown to directly stimulate nerves without any chemical pretreatment or genetic alteration.1–8 Likewise, IR pulse exposure has also been demonstrated to block action potential (AP) generation and propagation.9–13 While a rapid increase in temperature, due to absorption of the laser radiation, is required to evoke the neural depolarization, and IR stimulation pulses have been shown to produce an acoustic pressure wave,14–17 the mechanism(s) to stimulate or inhibit an AP is not fully understood.18 Certain thermal and mechanical mechanisms17,19 involving ion channels, such as transient receptor potential (TRP) channel activation,20 plasma membrane poration,21 and/or membrane potential changes, are suggested as explanations for IR neural stimulation and inhibition, together termed IR neural modulation (INM).1 Shapiro et al.22,23 also showed that the rapid temperature change slightly depolarizes the plasma membrane through capacitive charging. This effect could initiate AP firing in neurons but cannot explain IR-induced neuronal inhibition.
While much research into the mechanisms underlying IR stimulation has focused on the interaction of the IR pulse with plasma membrane, the diverse responses to INM suggest the possibility that intracellular physiological regulatory and compensatory mechanisms are involved in observed cell behavior. A critical role of intracellular regulation in cellular stimulation from thermal gradients has been indicated in several cell types. In HeLa cells, thermal rises of only a few tenths degrees, but 1- to 2-s long, have been shown to create a slight uptake of by sarco/endoplasmic reticulum (SERCA) and then an overshoot of cytoplasmic free from the ER due to -channels activation.24 Additionally, IR-induced intracellular transients originating from mitochondrial stores have been shown to be sufficient to modulate the activity of excitable neonatal cardiomyocytes, spiral and vestibular ganglion neurons.25,26 However, the addition of endoplasmic ryanodine receptors (RyR) blockers significantly reduced IR-induced response as well. IR pulses could also produce contraction of cardiomyocytes in the -free media and without noticeable transients.27,28 These results suggest that internal modulatory mechanisms might dominate over influx during IR stimulation.
Recently, we demonstrated that in nonexcitable CHO cells, a IR pulse exposure initiates the phosphatidylinositol4,5-biphosphate () intracellular signaling cascade.21 This critical physiological regulatory mechanism culminates in production of multiple second messengers, including -dependent intracellular release and activation of -dependent phospholipase C (PLC) and protein kinase C (PKC).29–31 Intracellular activity of PKC has been implicated in the modulation of thermo-sensitive TRP channels (TRPV1-4, TRPM8, and TRPA1).32,33 signaling is also involved in regulation and sensitization of the store-operated TRP channels (SOC),34–37 some of which are reported to be the core of the mechanosensitive system of mammalian cells.38–40 Neuronal voltage-gated channels (VGCC), SOC, thermo- and mechanosensitive TRP channels all transport into the cells and could be responsible for IR-induced intracellular increase as an alternative to possible plasma membrane nanoporation.21 also plays multiple roles in cellular physiology, including acting as a charge carrier across the plasma membrane and as a second messenger itself, enabling additional modulatory mechanisms. Thus, it is not surprising that intracellular fluctuations are accepted as one of the main hallmarks of neuronal excitability and could be a critical component for understanding the mechanisms of IR-induced neurological stimulation or inhibition.
In this paper, we provide data to progress the fundamental understanding of IR modulation of neurons by revealing the dependence of IR-induced mobilization on activation of intracellular stores and itself, whether from an internal or extracellular origin. By using ratiometric calcium imaging, we obtain quantitative measurements of calcium concentration to limit potential complications of intensity-based calcium indicators in environments with changing baseline cytosolic concentrations. Since mitochondrial cycling is important in regulation of homeostasis of all mammalian cells, we also use the innate difference in stores between nonexcitable and excitable (neuron-derived) cell types to compare the sensitivity of IR-induced response to these stores. , a nonexcitable cell line that lacks VGCCs,41 and rodent NG108 neuroblastoma, a neuro-derived cell line that does not produce AP in an early undifferentiated state but does contain multiple voltage-gated channels,42 were used to directly compare the sensitivity of IR-induced response without confounding effects from AP.
2.
Materials and Methods
2.1.
Cell Culture
Rodent neuroblastoma-glioma cells (NG108) were grown in Dulbecco’s modified Eagle’s medium without sodium pyruvate containing 10% fetal bovine serum, penicillin, streptomycin, 0.1 mM hypoxanthine, 400 nM aminopterin, and 0.016 mM thymidine. Chinese hamster ovarian cells () stably expressing human muscarinic acetylcholine receptor type 1 () were grown in F-12K medium containing 10% fetal bovine serum, penicillin, and streptomycin. Geneticin® (G418) is used in the CHO medium to maintain the expressing phenotype. Both cell lines were cultured at 37°C, 5% , and 95% humidity.
2.2.
Solutions
Solutions were exchanged through bath application using a Warner Instruments perfusion system at a flow rate of . Unless otherwise noted, in most experiments, we used a standard external buffer solution (pH 7.4, 290 to 310 mOsm) that consisted of 2 mM magnesium chloride (), 5 mM potassium chloride (KCL), 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), 10 mM glucose, 2 mM calcium chloride (), and 135 mM sodium chloride (NaCl). In some experiments (which are noted in the text), the was replaced with 2 mM Na-ethylene glycol-bis(-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) to create -free external buffer.
To investigate the sources of IR-induced intracellular rises and compare IR effects with well-known effects caused by endogenous PLC activation, some experiments were paired with -coupled receptor agonist, oxotremorine (OxoM, ), -coupled receptor agonist, bradykinin (BK, 100 nM), or RyR agonist caffeine (10 mM). Additionally, we used receptor () blockers xestospongin C (XeC ), 2-aminoethoxydiphenil borate (2-APB ), and RyR blocker ryanodine (). Media, chemicals, and pharmaceuticals were obtained from Life Technologies, Tocris Bioscience, or Sigma-Aldrich. In initial experiments, propidium iodide (PI) (BD Bioscience) was added to the external solution to a concentration of to verify cell viability43 and safe IR fiber placement.
2.3.
Infrared Laser Stimulation
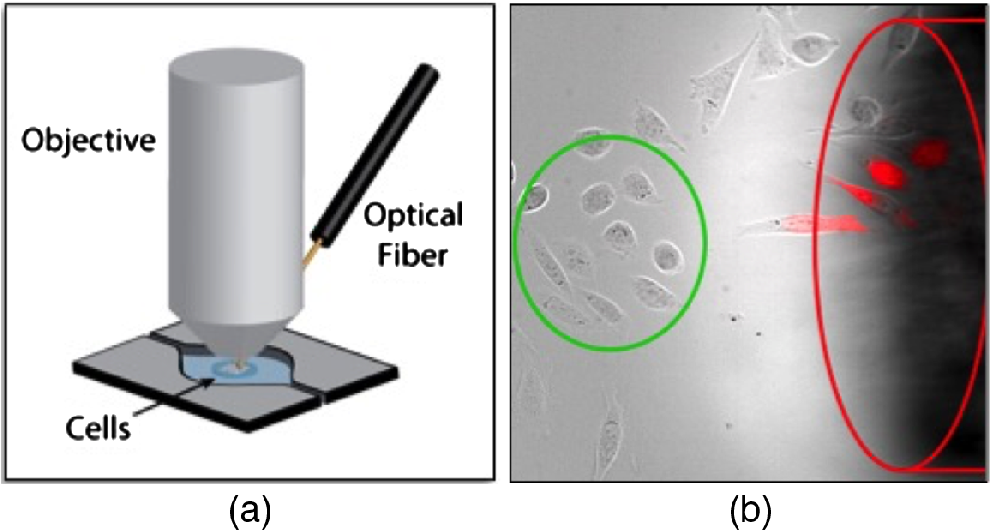
An Acculight Capella IR diode laser (Lockheed Martin) with a center wavelength of 1869 nm was used to stimulate the cells. As demonstrated in Fig. 1(a), the laser light was delivered to the sample by a core optical fiber. A region in the center of the fluorescent image was used to analyze the response, to ensure uniformity of exposure [Fig. 1(b), green circle]. The fiber tip (top edge) was positioned by a micromanipulator about away from the center to avoid obstruction of the region of interest by the fiber. The laser pulse was synchronized with the microscope using the Olympus real-time controller. The rapid rise temperature during stimulation caused intensity fluctuations in the images, possibly from thermal lensing. This “spiking” artifact effect was manually removed from data sets for clear presentation of IR-induced intracellular changes.
Fig. 1
(a) Diagram showing the position of the IR fiber in relation to the sample and (b) actual image of the cells and optical fiber. Cells in the green circle were used for measurements. The delivery fiber is outlined in red. PI is shown as the red fluorescence signal overlay. (From Olsovsky et al.46)
All IR stimulation experiments were performed with the laser set to deliver 5 pulses (a 1-s, 5-Hz pulse train) with individual pulse durations from 2 (2.5 mJ) to 3 ms (3.8 mJ). Pulse energy was determined at the fiber and the absorption of water was not taken into account. To ensure that the IR laser pulse was not acutely damaging the cells, uptake of PI was monitored after IR pulse exposure. Uptake of PI can indicate damage to the plasma membrane and PI was seen in cells were directly beneath and in front of the fiber where the temperature rises were significantly higher [Fig. 1(b)]. Thus, the cells that were used for experiments [Fig. 1(b), green circle] were selected from a region that did not demonstrate any PI uptake after many minutes after the IR exposure.
2.4.
Measurement of Calcium
Cells were plated on poly-L-lysine coated glass coverslips and kept in a 37°C, humidified (5% ) incubator for 24 to 48 h before imaging. The cells were then loaded with Fura-2 probe in a standard external buffer solution containing Fura-2 and 0.05% pluronic acid at 20°C for 30 min. The dye solution was then replaced with standard outside buffer solution for at least 15 min before imaging.
Fluorescent images were recorded using an Olympus epi-fluorescence microscope with a Lambda DG arc lamp and filter, a Hamamatsu Orca Flash 4.0 sCMOS camera, and an Olympus real-time controller. The real-time controller synchronizes the Lambda filter and camera so that an image using 340-nm excitation wavelength is captured immediately before another image using 380-nm excitation wavelength. The two images are compiled into a ratiometric image. The measured background and average autofluorescence for each cell line were subtracted before calculating the ratio. This ratio correlates to the concentration of calcium and is less vulnerable to artifact caused by variations in intensity due to, for example, defocus or sample thickness. The ratios were converted to concentrations using the following equation:
where is the measured ratio from the image and is the dissociation constant of Fura-2 as reported by Grynkiewicz et al.44 , , and are the minimum ratio, maximum ratio, and scaling factor, respectively, obtained by using Fura-2 calibration kit from Invitrogen. The calibration kit samples were pH 7.2, ionic strength 100 mM KCl, and Fura-2. is the measured ratio from the images of the sample containing free calcium and is the ratio from the images of the sample containing free (beyond saturation of Fura-2). is the fluorescence using 380-nm excitation on the free sample over the fluorescence from the free sample. The final values used in our experiments for , , , and were 224 nM, 0.207, 7.18, and 7.5, respectively.
3.
Results and Discussion
3.1.
Intracellular Ca2+ After Infrared Exposure
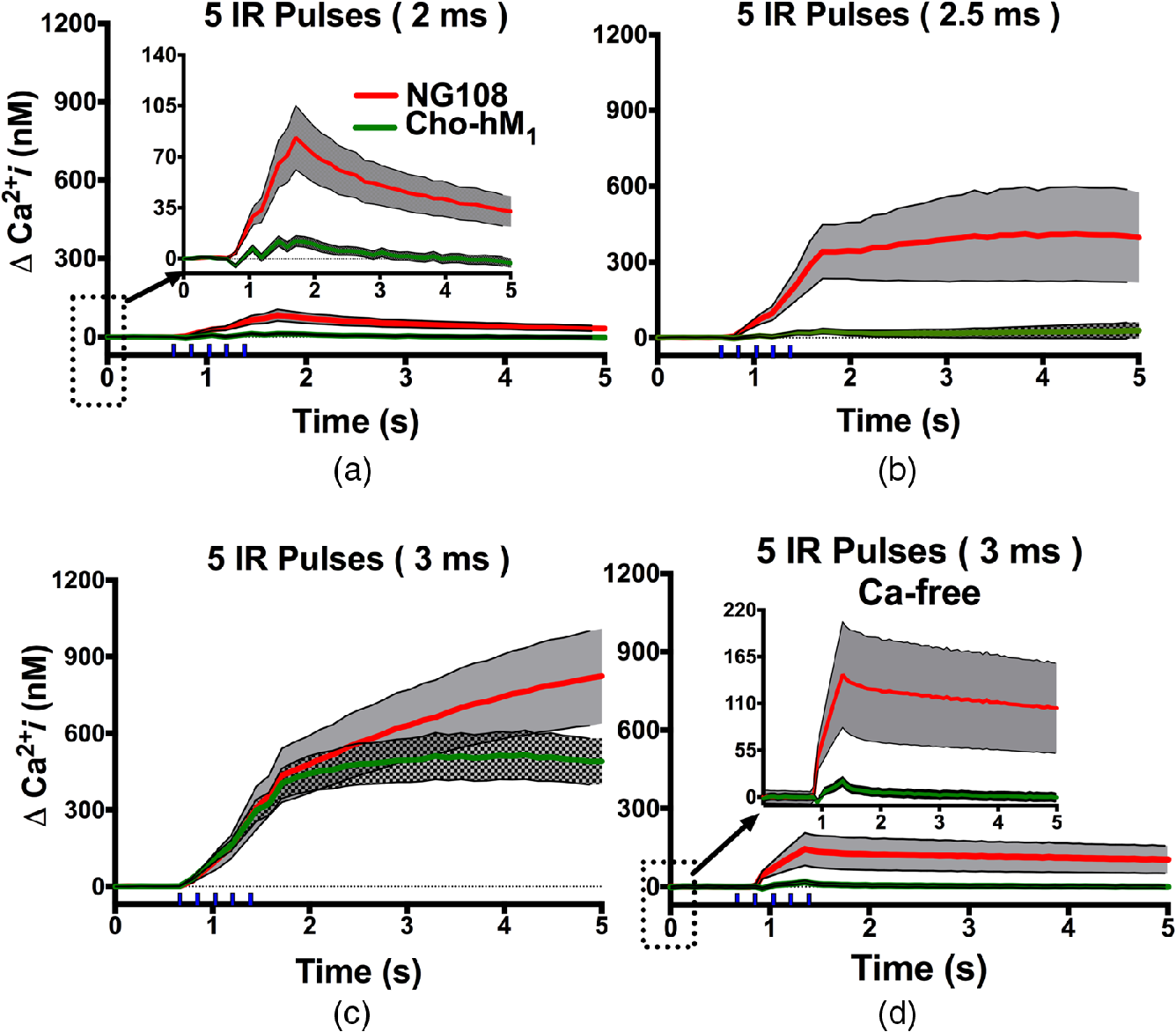
The resulting traces for the intracellular concentration increase after IR pulse exposures are shown in Fig. 2. A train of five IR pulses of 2, 2.5, or 3 ms duration started 660 ms after the beginning of image acquisition and lasted for 800 ms. In the -containing standard outside buffer solution, noticeable intracellular increases appeared during IR pulses and peaked 260 ms after the train in both NG108 and . rise began immediately after the first IR pulse and increased with each subsequent pulse and has previously been shown to be evoked by each laser pulse.45 The mean amplitudes at the peak of intracellular after 2 and 2.5 ms IR trains were significantly lower in than in NG108 [Figs. 2(a) and 2(b)]. The delta changes were () versus () for 2 ms IR pulse trials and () versus () for 2.5 ms pulses for versus NG108, respectively (, unpaired two-tailed -test). For the longer pulses [Fig. 2(c), 3 ms, 3.8 mJ], the intracellular increases were much higher than at lower IR pulses amplitudes, but increases observed between and NG108 become statistically insignificant (, versus , , respectively, ). Additionally, intracellular rise reaches a plateau 1.5 s after the end of the pulse train in CHO-hM1 but continues to rise in NG108 exposed cells. We then compared the increase in intracellular after a train of 3 ms pulses in -chelated extracellular media [Fig. 2(d)]. We found these intracellular concentration increases to be much smaller than in experiments with -containing external solution (postexposure , for and , for NG108), but still determined significant.
Fig. 2
Comparison of intracellular increases after train of IR pulses of different duration between NG108 and cell lines. (a–c) Exposures were performed in containing extracellular media. (d) Experiments performed in chelated outside media. Error bars (gray area) represent the standard error (SE) of the mean of 5 to 32 cells per group. Vertical ticks above -axis indicate the IR pulses train.
From our previous work suggesting that rises from IR pulse exposure were due to influx from extracellular media,21,46 we hypothesized that the exposure may create small pores in the plasma membrane. However, the presence of an intracellular rise in the absence of external suggests that increases after IR exposure is not the result of simple passive diffusion through a permeabilized plasma membrane, but rather, a complex and regulated process, possibly through the involvement of -sensitive endoplasmic reticulum (ER) stores and -induced--release (CICR) from ryanodine-sensitive stores. Additionally, in both -containing and -chelated solution, did not exhibit as high a increase as NG108. The composition and distribution of plasma membrane ion channels responsible for normal cellular homeostasis and function are markedly different between mammalian excitable and nonexcitable cells. Similar differences are also present in the membranes of major intracellular stores. For example, muscular, neuronal, and cardiomyocyte cells widely express both RyR and in the sarco-ER, but nonexcitable cells express mostly intracellular .47 The differences in expression in the ER of RyR and in excitable and nonexcitable cells could be one of the main regulatory mechanisms responsible for the sensitivity of these cells to external stressors, such as IR stimulation.
Furthermore, the rise can be blocked in IR-exposed NG108 and in -chelated external media supplemented with thapsigargin.46 Thapsigargin blocks SERCA, which normally pumps from the cytosol into the lumen of the sarco-ER,48–50 thereby resulting in the depletion of intracellular stores. Remaining is eventually cleared by plasma membrane pumps.51 Thus, the lack of a increase after IR stimulation seen in these depleted cells suggests that increase in -free solution may originate from ER or ryanodine-sensitive stores.
3.2.
Role of Intracellular Ca2+ Stores in Ca2+ Rises After Infrared Exposure
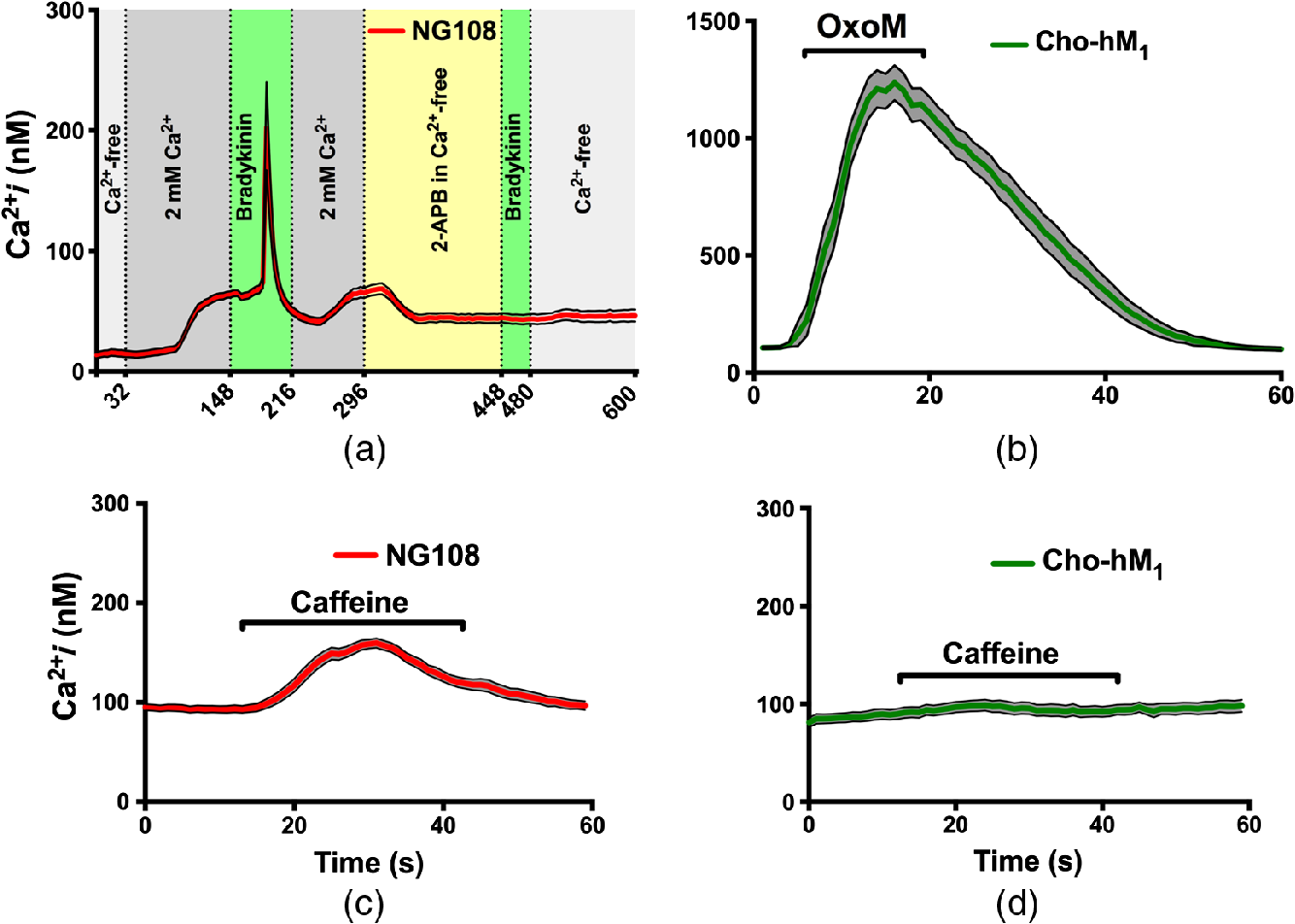
To investigate the role that these stores may be playing in the INM response, we then performed a series of experiments with agonists of the RyR and in NG108 and . First, to demonstrate the functional expression of intracellular ER receptors and capability of NG108 to adjust to changes in extracellular , a series of solution changes were conducted during imaging (Fig. 3). NG108 were bathed in -chelated buffer for 30 min before beginning ratiometric imaging to partially deplete intracellular out of the unstimulated cells.52 The normal resting intracellular concentration is between 50 and 100 nM,53,54 but this exposure depleted it to [Fig 3(a)]. Shortly after beginning perfusion of cells with -containing solution, the resting intracellular concentration reached a normal due to a capacitive entry mechanism. Treatment of NG108 with 100 nM BK peptide caused activation of the -coupled receptors, consequentially initiating signaling and production of the . After the -induced spike, we applied 2-APB () in -chelated buffer to block and slightly deplete intracellular stores.55 This manipulation prevented the -induced spike after secondary application of the BK and confirmed the functional role of in NG108. Similarly, stably expresses the -coupled receptors, so application of a high concentration of agonist OxoM () resulted in a strong -dependent response [Fig. 3(b)].29 To demonstrate CICR from ryanodine stores, we applied caffeine (10 mM) to sensitize RyR and allowed basal cytosolic calcium levels to actuate CICR.56,57 A cytoplasmic rise can be seen in the NG108, but , which do not contain ryanodine stores, shows no response [Figs. 3(c) and 3(d)].
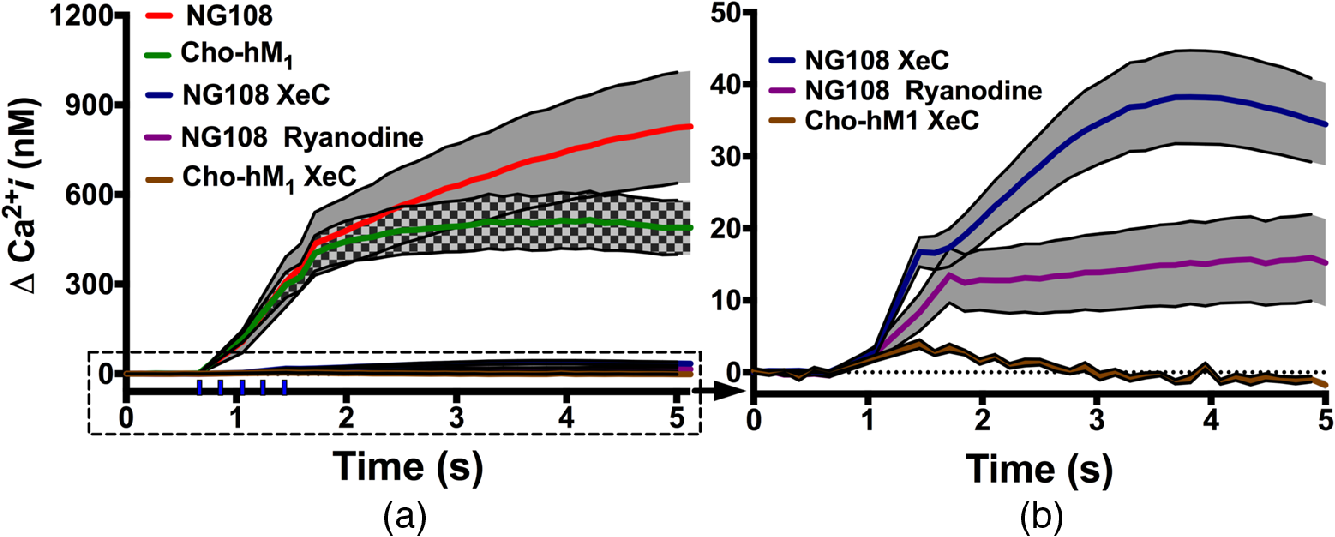
To investigate the role of RyR and in the IR-induced changes in intracellular dynamics, we exposed both NG108 and to a 3-ms IR pulses train in the presence of several receptor antagonists. Antagonists of RyR and dramatically reduced IR-induced intracellular response in both cell lines [Fig. 4(a)], suggesting that physiological regulatory mechanisms are predominate in the cellular response to IR stimulation.
Fig. 3
Intracellular RyR and in the NG108 and cells. (a) Demonstration of the NG108 cells () capability to adjust to changes in extracellular concentration and respond to 100 nM BK-induced activation. (b) OxoM ()-induced intracellular rise in cells () due to ER activation. (c) Caffeine (10 mM)-induced increase due to intracellular RyR receptors activation in the NG108 cells (). (d) Lack of response to 10 mM caffeine in CHO-hM1 cells (). Error bars (black outline with gray area fill) represent the SE of the mean.
Fig. 4
NG108 and responses in -containing outside buffer after trains of 3-ms duration IR pulses with and without intracellular RyR and blockers. (a) IR-induced changes in intracellular dynamics. The traces without antagonists are the same as in Fig. 2 and presented here for comparison. Vertical ticks above -axis indicate the IR pulses train. (b) Magnification of the responses with RyR and antagonists. Error bars (black outline with gray or black/gray checked pattern fill areas) represent the SE of the mean ( to 16).
The stores, as shown above (Fig. 4), are present in both NG108 and . We pretreated cells for 20 min with XeC (), a specific inhibitor of the -dependent release.58 In , XeC nearly completely blocked the rise in intracellular levels (postexposure , ), even in -containing outside buffer. Despite the fact that such a small response is within normal intracellular physiological fluctuations, the rise correlates temporally with IR exposure [Fig. 4(b)]. This small increase could be due to capacitive entry of extracellular through diacylglycerol (DAG)-sensitive TRP/SOC channels or from incomplete block of the .34,35,59,60 In NG108, a similar small intracellular response could lead to CICR from RyR stores. Indeed, IR stimulation of NG108 cells in -chelated outside buffer and treated with XeC () resulted in a small, but significant rise (postexposure , ). Additionally, NG108 pretreated with RyR antagonist ryanodine61 () in -containing buffer, showed a large reduction in rise after IR stimulation (postexposure , ), with the small rise in possibly resulting from stores or capacitive entry37,40 without CICR [Fig. 4(b)]. These results show that stores are involved in signaling from INM in both cell lines but are not the sole source in NG108.
Our observations further suggest that differences between excitable and nonexcitable cells in IR-induced responses could be due to distinct expression of intracellular RyR and in the ER of these cells. RyR and have been shown to be activated in parallel with store-operated entry (SOCE) and strongly contribute to the global response.62 Depletion of these intracellular stores can initiate SOCE through plasma membrane SOC channels. Thus, much of the observed increase in the NG108 could be due to direct or indirect activation of RyR and additional to SOCE intracellular regulatory mechanism. In NG108, it has been demonstrated that depolarization-induced entry evoked CICR only from the ryanodine-sensitive stores,63 which greatly contribute to general response.
Previous experiments on HeLa cells, cardiomyocytes, and neurons have demonstrated the critical role of intracellular regulation in thermal gradient stimulation mechanisms. In HeLa cells, during second-long heating of , a decrease in was observed, theorized to be due to an increase of SERCA activity along with a decrease in the open probability of the ER and RyR. After the exposure, the rapid cooling was hypothesized to increase the open probability of these ER conducting channels, leading to an overshoot of cytoplasmic . This IR-induced uptake by SERCAs and its asymmetrical outflow via intracellular ER were proposed as a general mechanism of the temperature-dependent changes in dynamics.24 While we did not observe a decrease in in these experiments, due to the brevity of our pulses and experimental parameters, this hypothesized sensitivity of the ER could contributed the overshoot observed from IR pulses. IR rapid heating/cooling of water also creates capacitive photothermal currents, which results in plasma membrane depolarization/repolarization19 and thus possible activation of the voltage sensitive phosphatase (Ci-VSP). Recently, Ci-VSP was shown to regulate signaling in the plasma membrane64–66 and could be accounted for the initial depletion during IR-induced cellular response.
Previous IR pulse experiments in cardiomyocytes and spiral and vestibular ganglion neurons indicated that the calcium signaling originated from the mitochondria. However, three (ryanodine, cyclopiazonic acid, and ruthenium red) of the pharmaceutical compounds used in these studies have direct severe inhibitory effect on the RyR and ER, indicating a likely critical importance of internal ER pools/receptors in IR-induced INM in addition to alteration of mitochondrial function.26 By using two cell lines with innate differences in ER receptors, we demonstrate the role that the interplay between these two receptors has on the response the IR exposure.
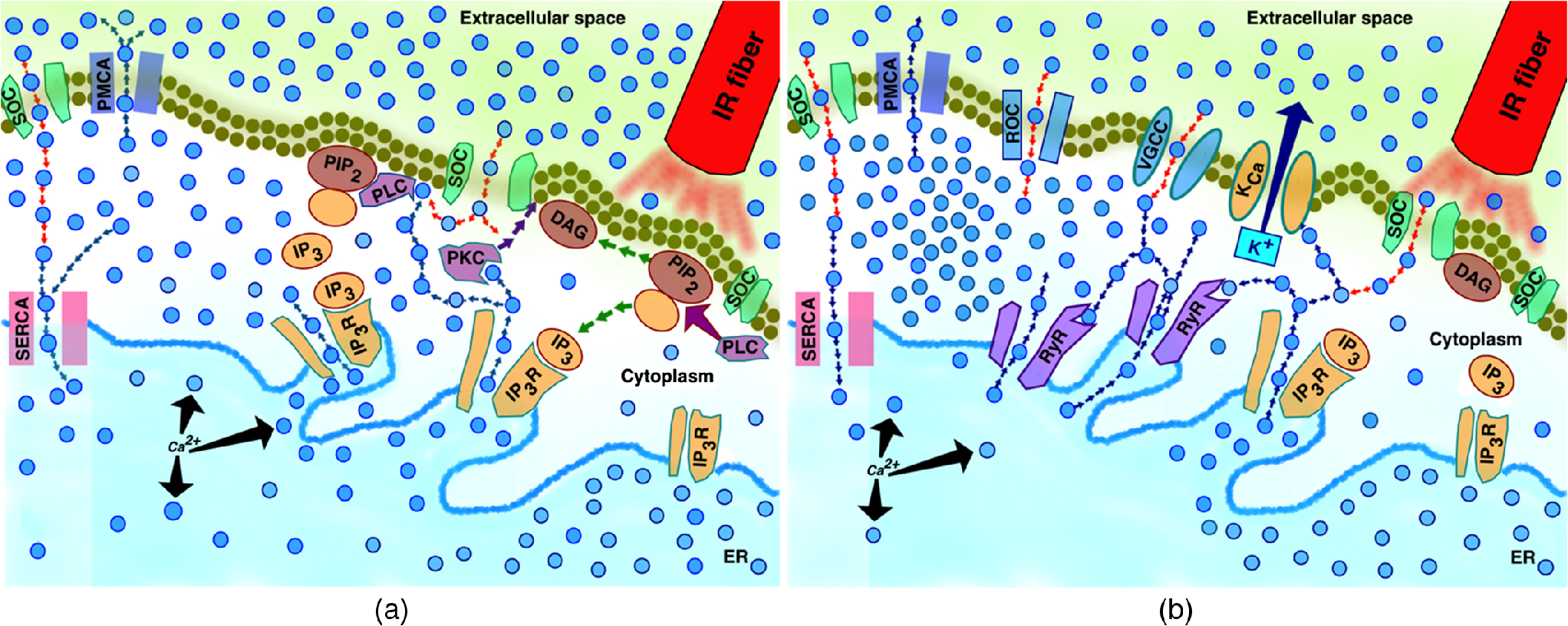
Previously, we found that IR pulses initiated the intracellular phosphoinositide signaling cascade in .21 This response appeared similar to one initiated by activation of -coupled receptors and resulted in production with possible consequential depletion of the intracellular ER stores. is a main component of the intracellular calcium signaling and provides a direct link between cellular plasma membrane and prime intracellular store, the ER.30,67–70 The exact mechanism of IR-induced activation of signaling is unknown, but hypothetical schematics of the IR-induced responses are presented in Fig. 5.
Fig. 5
Simplified hypothetical schematic of IR-induced response between (a) nonexcitable and (b) excitable cells.
In nonexcitable cells [Fig. 5(a)], IR-induced PLC-dependent hydrolysis or depletion leads to production of and DAG (green arrows). DAG and its derivative, arachidonic acid, activate -conducting TRP SOC channels34,35,40,60 and initiates intracellular release through activation of on the ER (blue arrows out of ER). Intracellular activates cytoplasmic PKC, which has a high affinity to DAG.71,72 Active PKC translocates toward DAG (purple arrows) and phosphorylates TRP channels, keeping them in the open state longer.73 Extracellular started to influx into cytosol through TRP/SOC channels due to the SOCE mechanism (red arrows). High levels of intracellular catalyze PLC activity, leading to stronger hydrolysis, and potentiating the reaction described above.29,74 High intracellular is eventually pumped out of the cell by plasma membrane -ATPase and into ER stores by SERCA.75,76 In excitable, specifically neuronal cells [Fig. 5(b)], in addition to the reactions described above and SOCE, the intracellular increase is achieved through additional mechanisms, including strong sensitization of neurons by and -activated ryanodine-sensitive release.63,77–79 The interplay of these two intracellular pools is critically important, since it leads to much stronger phenotypic response. - and -dependent modulation of the neuronal potassium channels leads to changes in membrane potential and depolarization.30,80 As a consequence of depolarization and activation of the VGCC, influx could also evoke CICR through RyR receptors.63,79 Last, the overall neuronal activity induces influx through excitatory neurotransmitters and receptor-operated channels.81–84 Therefore, IR-induced changes of intracellular signaling and dynamics in neurons can explain both stimulation and modulation mechanisms. While additional studies are needed, our experiments presented here indicate that intracellular in the ER play an important role in both excitable and nonexcitable cell lines, with the IR-induced response augmented by RyR in excitable cells, thus strongly reinforcing our hypothesis.
4.
Conclusions
This study directly compared mobilization in two very different cells lines, neuronal-like NG108 and epithelial , to determine the source of rise resulting from INM. As both NG108 and cell models demonstrate an increase in intracellular after IR stimulation, the results suggest that influx from extracellular space is accompanied by derived from the intracellular and ryanodine-sensitive stores. However, the intracellular response in NG108 cells was determined significantly greater, suggesting that interplay of and ryanodine intracellular pools is critically important to augment the rise through CICR after an IR pulsed exposure event.
Disclosures
The authors have no additional relevant financial interests or potential conflicts of interest.
Acknowledgments
This work was supported by the Air Force Office of Scientific Research (LRIR #15RHCOR204). Support for Mr. Cory A. Olsovsky was provided by a Repperger Research Intern Program administered by the Air Force Research Laboratory, 711th Human Performance Wing. cells were donated by Dr. Mark S. Shapiro (University of Texas Health Science Center at San Antonio, Department of Physiology).
S. A. Shintani et al.,
“High-frequency sarcomeric auto-oscillations induced by heating in living neonatal cardiomyocytes of the rat,”
Biochem. Biophys. Res. Commun., 457
(2), 165
–170
(2015). http://dx.doi.org/10.1016/j.bbrc.2014.12.077Google Scholar
29.
L. F. Horowitz et al.,
“Phospholipase C in living cells activation, inhibition, requirement, and regulation of M current,”
J. Gen. Physiol., 126
(3), 243
–262
(2005). http://dx.doi.org/10.1085/jgp.200509309 JGPLAD 0022-1295 Google Scholar
30.
N. Gamper and M. S. Shapiro,
“Regulation of ion transport proteins by membrane phosphoinositides,”
Nat. Rev. Neurosci., 8
(12), 921
–934
(2007). http://dx.doi.org/10.1038/nrn2257Google Scholar
S. Mandadi, P. J. Armati and B. D. Roufogalis,
“Protein kinase C modulation of thermo-sensitive transient receptor potential channels: implications for pain signaling,”
J. Natl. Sci. Biol. Med., 2
(1), 13
–25
(2011). http://dx.doi.org/10.4103/0976-9668.82311Google Scholar
33.
S. Mandadi et al.,
“Activation of protein kinase C reverses capsaicin-induced calcium-dependent desensitization of TRPV1 ion channels,”
Cell Calcium, 35
(5), 471
–478
(2004). http://dx.doi.org/10.1016/j.ceca.2003.11.003 CECADV 0143-4160 Google Scholar
34.
I. Jardin et al.,
“Phosphatidylinositol 4, 5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets,”
Biochim. Biophys. Acta, 1783
(1), 84
–97
(2008). http://dx.doi.org/10.1016/j.bbamcr.2007.07.007Google Scholar
G. Grynkiewicz, M. Poenie and R. Y. Tsien,
“A new generation of indicators with greatly improved fluorescence properties,”
J. Biol. Chem., 260
(6), 3440
–3450
(1985). Google Scholar
C. A. Olsovsky et al.,
“Origins of intracellular calcium mobilization evoked by infrared laser stimulation,”
Proc. SPIE, 9321 93210L
(2015). http://dx.doi.org/10.1117/12.2079895 PSISDG 0277-786X Google Scholar
G. Inesi et al.,
“Cell-specific promoter in adenovirus vector for transgenic expression of SERCA1 ATPase in cardiac myocytes,”
Am. J. Physiol., 274
(Pt. 3), C645
–C653
(1998). Google Scholar
49.
M. S. Kirby et al.,
“Thapsigargin inhibits contraction and transient in cardiac cells by specific inhibition of the sarcoplasmic reticulum pump,”
J. Biol. Chem., 267
(18), 12545
–12551
(1992). Google Scholar
R. ChakrabartiR. Chakrabarti,
“Calcium signaling in non-excitable cells: release and influx are independent events linked to two plasma membrane entry channels,”
J. Cell Biochem., 99
(6), 1503
–1516
(2006). http://dx.doi.org/10.1002/(ISSN)1097-4644Google Scholar
53.
J. Tong, T. V. McCarthy and D. H. MacLennan,
“Measurement of resting cytosolic concentrations and store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant release channels,”
J. Biol. Chem., 274
(2), 693
–702
(1999). http://dx.doi.org/10.1074/jbc.274.2.693Google Scholar
54.
J. L. Nunez and M. M. McCarthy,
“Resting intracellular calcium concentration, depolarizing gamma-aminobutyric acid and possible role of local estradiol synthesis in the developing male and female hippocampus,”
Neuroscience, 158
(2), 623
–634
(2009). http://dx.doi.org/10.1016/j.neuroscience.2008.09.061Google Scholar
I. Ricard et al.,
“A caffeine/ryanodine-sensitive pool is involved in triggering spontaneous variations of in Jurkat T lymphocytes by a Ca(2+)-induced release (CICR) mechanism,”
Cell Signal, 9
(2), 197
–206
(1997). http://dx.doi.org/10.1016/S0898-6568(96)00141-6Google Scholar
L. Lemonnier, M. Trebak and Jr J. W. Putney,
“Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4, 5-bisphosphate,”
Cell Calcium, 43
(5), 506
–514
(2008). http://dx.doi.org/10.1016/j.ceca.2007.09.001 CECADV 0143-4160 Google Scholar
A. M. Vites and A. J. Pappano,
“Distinct modes of inhibition by ruthenium red and ryanodine of calcium-induced calcium release in avian atrium,”
J. Pharmacol. Exp. Ther., 268
(3), 1476
–1484
(1994). Google Scholar
62.
S. Dadsetan et al.,
“Store-operated influx causes release from the intracellular channels that is required for T cell activation,”
J. Biol. Chem., 283
(18), 12512
–12519
(2008). http://dx.doi.org/10.1074/jbc.M709330200Google Scholar
63.
P. Ronde, J. J. Dougherty and R. A. Nichols,
“Functional IP3- and ryanodine-sensitive calcium stores in presynaptic varicosities of NG108-15 (rodent neuroblastoma x glioma hybrid) cells,”
J. Physiol., 529
(Pt. 2), 307
–319
(2000). http://dx.doi.org/10.1111/tjp.2000.529.issue-2Google Scholar
Y. Murata and Y. Okamura,
“Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2,”
J. Physiol., 583
(Pt. 3), 875
–889
(2007). http://dx.doi.org/10.1113/jphysiol.2007.134775Google Scholar
M. J. Berridge, P. Lipp and M. D. Bootman,
“The versatility and universality of calcium signalling,”
Nat. Rev. Mol. Cell Biol., 1
(1), 11
–21
(2000). http://dx.doi.org/10.1038/35036035Google Scholar
70.
E. A. Finch and G. J. Augustine,
“Local calcium signalling by inositol-1, 4, 5-trisphosphate in Purkinje cell dendrites,”
Nature, 396
(6713), 753
–756
(1998). http://dx.doi.org/10.1038/25541Google Scholar
E. Oancea et al.,
“Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells,”
J. Cell Biol., 140
(3), 485
–498
(1998). http://dx.doi.org/10.1083/jcb.140.3.485Google Scholar
M. Seth et al.,
“Sarco(endo)plasmic reticulum ATPase (SERCA) gene silencing and remodeling of the signaling mechanism in cardiac myocytes,”
Proc. Natl. Acad. Sci. U. S. A., 101
(47), 16683
–16688
(2004). http://dx.doi.org/10.1073/pnas.0407537101Google Scholar
A. Sanchez-Perez et al.,
“Modulation of NMDA receptors in the cerebellum. II. Signaling pathways and physiological modulators regulating NMDA receptor function,”
Cerebellum, 4
(3), 162
–170
(2005). http://dx.doi.org/10.1080/14734220510008003Google Scholar
83.
R. Schneggenburger, F. Tempia and A. Konnerth,
“Glutamate- and AMPA-mediated calcium influx through glutamate receptor channels in medial septal neurons,”
Neuropharmacology, 32
(11), 1221
–1228
(1993). http://dx.doi.org/10.1016/0028-3908(93)90016-V NEPHBW 0028-3908 Google Scholar
84.
D. M. Simeone, B. C. Kimball and M. W. Mulholland,
“Acetylcholine-induced calcium signaling associated with muscarinic receptor activation in cultured myenteric neurons,”
J. Am. Coll. Surg., 182
(6), 473
–481
(1996). Google Scholar
Biography
Gleb P. Tolstykh received the Presidential Research Fellowship award in 1995 and completed it at the University of Texas Health Science Center at San Antonio (USA). He continued his research career there, focusing on physiology and neuroscience. He was a Senior National Research Council Fellow (USA) from 2011 to 2013 at the Air Force Research Laboratory (AFRL). Currently, he is a principal scientist at General Dynamics Information Technology, investigating high-energy-induced effects on cellular homeostasis.
Cory A. Olsovsky is a graduate student in the Biomedical Engineering Department at Texas A&M University, College Station, Texas. He received his BS degree in biomedical engineering from Texas A&M University in 2011. His current research is focused on innovative techniques for confocal microscopy.
Bennett L. Ibey received his PhD in biomedical engineering from Texas A&M University in biomedical optics in 2006. He joined AFRL’s Radio Frequency Bioeffects Branch in 2007 and serves as a principal investigator for high peak power microwave bioeffects and nanosecond electric pulse research. He is an associate editor for the Bioelectromagnetics Journal and a lifetime member of SPIE.
Hope T. Beier has been a research biomedical engineer in AFRL’s Optical Radiation Bioeffects Branch since November 2012. She serves as a principal investigator for efforts using advanced optical techniques to investigate the effects of directed energy (laser and radio frequency) on biology. She received her PhD in biomedical engineering from Texas A&M University in 2009. She joined AFRL in 2010 as a National Research Council Postdoctoral Research Associate.
The alert did not successfully save. Please try again later.
Gleb P. Tolstykh, Cory A. Olsovsky, Bennett L. Ibey, Hope T. Beier, "Ryanodine and IP3 receptor-mediated calcium signaling play a pivotal role in neurological infrared laser modulation," Neurophoton. 4(2) 025001 (5 April 2017) https://doi.org/10.1117/1.NPh.4.2.025001