
|
|
1.IntroductionProteins have an intrinsic propensity to self-assemble into aggregates. The process is initiated by the formation of an aggregation-nucleus from the native monomers; this is a small oligomeric species consisting of only a few monomer units, from which aggregation then proceeds via association of further monomers. Kinetically, the nucleus is a unit for which the rate of further monomer association is larger than that of dissociation. Such oligomers are called “on-pathway” species, because their presence increases the propensity of the protein pool to aggregate. In contrast, oligomers that block the growth of aggregates are called “off-pathway” species; these can dissociate and act as a source for monomers.1 Usually, aggregation corresponds to the association of proteins, whose polypeptide chain is folded into a nonfunctional structural state; one says the protein is in a “misfolded state.” Aggregation thus propagates the misfolded protein state and this phenomenon is at the heart of a number of so-called “protein misfolding diseases.”2,3 A key problem in misfolding diseases is that normal protein homeostasis is disrupted in the cell, as aggregation lowers the propensity of the protein pool to remain soluble. Small oligomeric species may furthermore elicit toxicity directly through interference with vital metabolic processes in the cell. These problems are at the heart of major neurodegenerative diseases, such as Alzheimer’s Disease (AD) and Parkinson’s Disease (PD), in which usually harmless, soluble proteins self-assemble into highly regular, fibrillar aggregate structures called amyloids,4 which may elicit structure-specific phenotypes. In AD and PD, amyloids are formed from intrinsically disordered proteins (IDPs), i.e., those that normally lack defined secondary and tertiary structures under physiological conditions. In response to changes in the molecular environment or due to point mutations in their peptide sequence, IDPs partially fold and adopt metastable -sheet conformations (see Fig. 1). These partially folded states self-assemble into oligomers, which then gradually convert into stable, -sheet rich conformations, thus forming aggregation nuclei, from which elongation into the fibrillar amyloid state then proceeds through consecutive monomer addition.5 In both AD and PD, aggregation appears to be initiated in certain, well-defined regions in the brain, before the gradual spreading of aggregated species takes place throughout the entire brain. In the final stages of the disease, a significant fraction of the brain mass of affected patients is replaced by insoluble solid plaques, which are predominantly composed of fibrillar amyloids of the proteins tau and amyloid () in Alzheimer’s, and the protein -synuclein in the case of PD. Fig. 1Simplified pathway toward amyloid fibril formation. In the misfolded-state proteins have an increased propensity to oligomerize, via association of their metastable beta-sheet domains. These can convert into more stable beta-sheet states and the ensuing oligomers act as the nuclei for the subsequent elongation reaction, which leads to the formation of so-called protofibrils. The final amyloid fibril usually consists of a number or intertwined protofibrils. 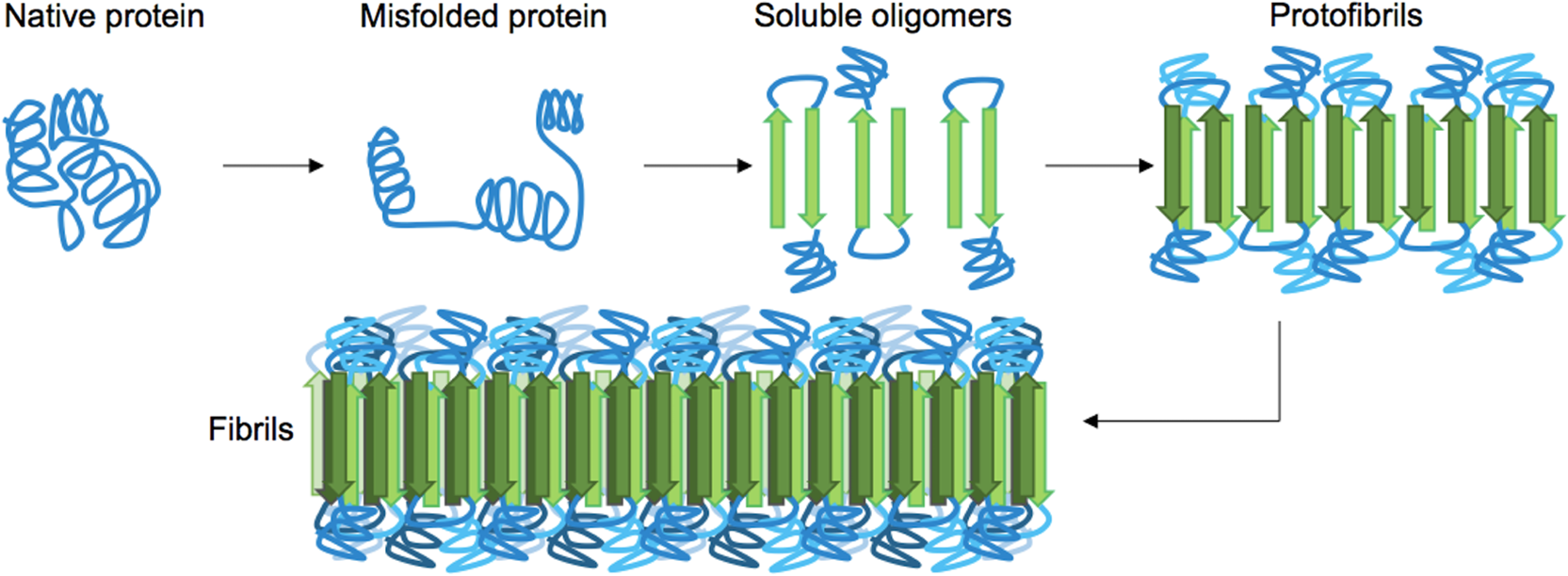 The environmental conditions that favor, or hamper, the self-assembly of proteins into amyloid fibrils, the propagation of misfolded species from cell to cell, the effect of point mutations on the aggregation process, the possible interaction with, and inhibition of, amyloid aggregation via small molecule drugs, the structural identification and physiological interaction of toxic species in brain cells, and so on are all questions at the heart of a better understanding of these devastating diseases and the focus of much ongoing biophysical research. Optical techniques play a crucial role in this effort, because of their nonintrusive nature, their suitability to track the kinetics of the self-assembly process in time and their high sensitivity and specificity in combination with fluorescent labeling techniques, which makes them suitable for applications in cells and even in vivo. In what follows, we review techniques pioneered in our laboratory and by others, for studies of amyloid formation optically, with nanoscale resolution. We begin with indirect methods, which we term “multiparameter imaging,” where molecular-level information is conveyed via changes in spectral properties of reporter fluorophores, e.g., changes in the fluorescence lifetime, or the polarization of emitted radiation. Bulk measurements with multiparameter methods are presented first, before applications are discussed in combination with microscopic imaging. We then review the application of single-molecule superresolution microscopy, which reveals morphological information of single aggregate clusters at the molecular scale. 2.Bulk Measurements to Study Fibril GrowthTraditional optical techniques well suited for bulk measurements include the use of far UV circular dichroism and Fourier transform infrared spectroscopy, which inform on the content of -sheet to -helical structure in unlabeled proteins.6,7 Furthermore, small molecule dyes, such as thioflavin T (ThT), that bind with a high propensity to the -sheet rich fibrillar state of amyloids can be used to track the aggregation state in bulk.8 The rise in ThT fluorescence emission in mixtures containing aggregating peptides is a popular method to track the kinetics of amyloid formation in vitro.9 Although the exact mechanisms by which ThT fluorescence is affected by fibril binding is still incompletely understood, it is thought that intercalation of the dye in the fibril grooves leads to a stabilization of the molecule in a configuration, which enhances electron delocalization and thus transition dipole moments. An intriguing phenomenon recently discovered is that the formation of amyloid fibrils is accompanied by the adoption of a structure-specific intrinsic fluorescence in the visible range, independent of the presence of aromatic residues in the polypeptide chain.10,11 This provides for a tool in the diagnostics of aggregation kinetics in vitro and has been used as a quantitative assay, e.g., to study the elongation reactions of -synuclein,12 see Figs. 2(a), 2(b), and 2(d). This study focused on the elongation of fibrillar template species in the presence of monomers of -synuclein, and clearly exponential growth of fibrils is evident as a function of incubation time. Growth rates recede gradually, as the monomer concentration becomes depleted during fibril growth. Fig. 2Intrinsic fluorescence informs on amyloid growth kinetics without the requirement for any extrinsic fluorescence labels. (a) Short, fibrillar seeds of -synuclein, shown in blue, serve as the template from which fibril elongation proceeds through addition of monomers, shown in red. The top panel to the right shows atomic force microscopy (AFM) images of short fibril seeds at the start of the reaction, at . Below is an image of the same sample at . Clearly, the elongation of the seeds into long amyloid fibrils has taken place. (b) Structural model of amyloid fibril, consisting of intertwined protofibrils. (c) Zoomed-in region indicated in b that shows the atomic arrangement between adjacent N- and C-termini from intertwined protofibrils. (d) Rise in intrinsic fluorescence from the amyloid fibrils in time. The blue points correspond to the elongation of -synuclein seeds in the presence of -synuclein monomer. The black points correspond to the addition of -synuclein monomers to the -synclein seed fibrils and confirm that the two species cannot “cross-seed” aggregation. The fluorescence measurements were performed with a diode laser emitting near 405 nm and signals were detected around 450 nm. Similar spectral characteristics were observed for hen egg white lysozyme,11 tau,12 and various mutants of amyloid beta.13 Figure reprinted with permission from Ref. 12, copyright 2014 Wiley [panels (a) and (d)] and Ref. 13, copyright 2016 American Chemical Society [panels (b) and (c)]. 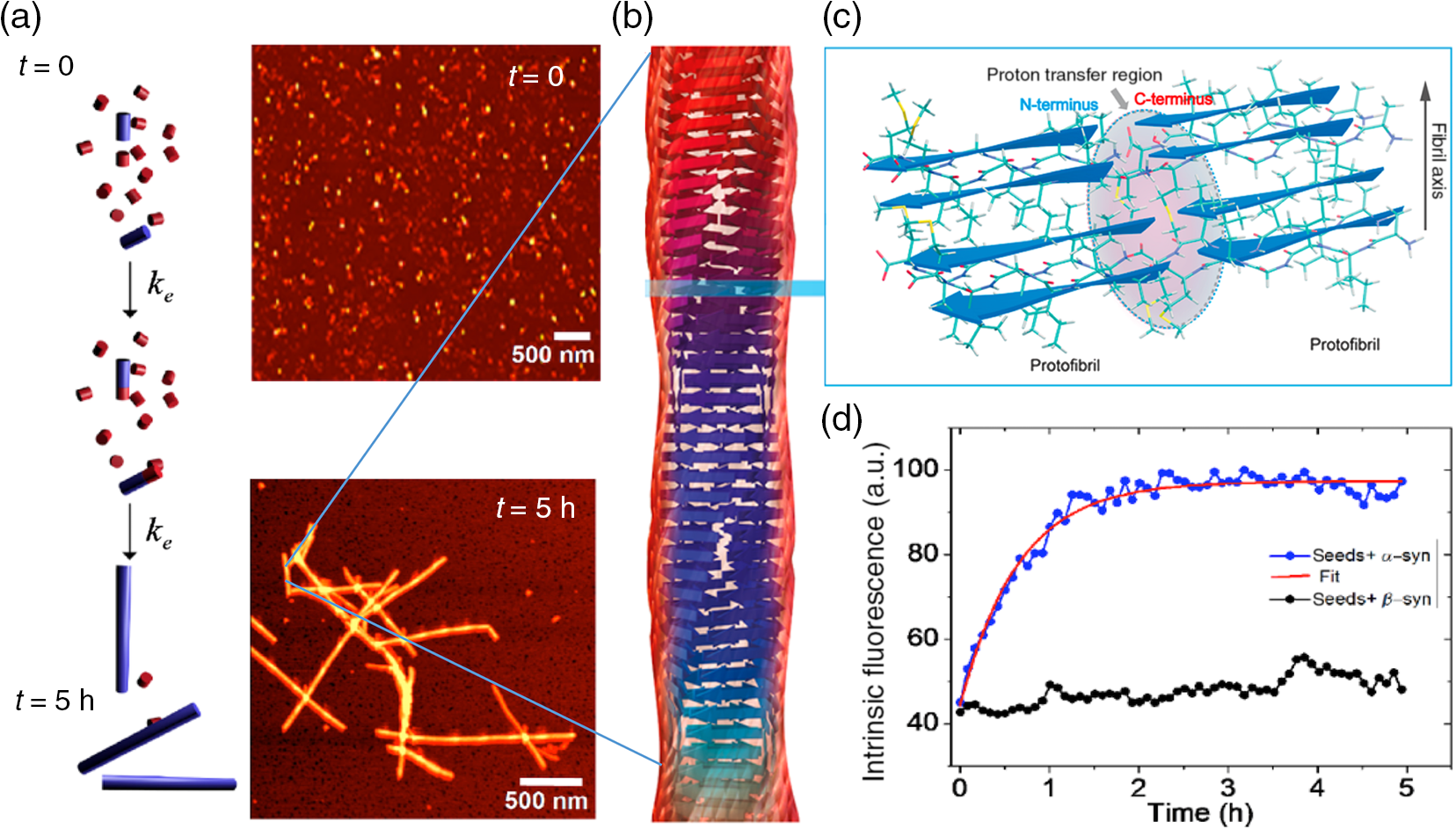 Fluorescence was furthermore confirmed to be independent of the presence of aromatic side chains in the studied proteins and to occur in a wavelength range far removed spectrally from that of traditional intrinsic protein fluorescence (excitation near 400 nm, emission emission range).12 The intrinsic fluorescence properties were found to be similar for many different amyloids tested and strong enough to render fibrils visible under the microscope. For example, absorption and emission spectra of the amyloid proteins tau, lysozyme, and amyloid beta were shown to exhibit very similar features to those of -synuclein, as reported in Refs. 10, 11, and 13, respectively. We have recently confirmed via ab initio molecular dynamics, and simulations that protons situated along H-bonds involving termini between adjacent fibril strands can be exceptionally labile. This leads to situations in which the proton can hop between the N- and C-termini of adjacent strands, a situation that is exacerbated via nuclear quantum effects. The result is a strong delocalization of protons along the H-bond co-ordinate axis, which in turn imparts a very significant effect on the optical properties of amyloid systems and likely also their physicochemical properties.13 While a useful tool for kinetic studies of purified protein in vitro, the intrinsic fluorescence is too unspecific and weak to be useful in more complex assays, e.g., for high-resolution microscopy techniques or for studies in cells or organisms. Specificity and sensitivity can be conferred via fluorescent labels that are covalently linked to the protein of interest. Labeling of primary amine groups is possible via succinimidyl esters, but control over the binding site can be difficult, causing possible interference with the amyloid self-assembly process.14 Site-specific labeling is possible via maleimide dyes, which react only with cysteine residues. Specific cysteine residues can be replaced or introduced through site selective mutagenesis; this allows the covalent and specific labeling of domains, which lie outside of the amyloid specific misfolding region, thus minimizing the risk of steric interference with the aggregation process.15 For superresolution microscopy in particular, this permits the introduction of highly efficient organic dyes, optimally suited to the imaging task at hand. In all cases, the propensity of the protein to aggregate with comparable behavior to the wild-type protein and to form fibrils featuring similar morphologies, needs to be carefully checked using biophysical assays, such as nuclear magnetic resonance, AFM, and scanning electron microscopy (SEM). This is particularly true for the use of fusion proteins as labels, whose size can exceed that of labeled peptide. Nevertheless, the use of variants of green fluorescent protein has been demonstrated successfully for a range of amyloid peptides, yielding similar aggregation kinetics and fibril morphology as the wild-type protein.16 3.Functional Imaging of Amyloid Growth KineticsFusion protein technology permits imaging directly at the live cell, or even organism, level.17,18 This opens a whole range of possibilities for functional imaging techniques, so-called multiparameter imaging techniques, that report on molecular self-assembly directly in aggregation models of patho-biological relevance. The use of homoFRET reports on aggregation via an increasing loss of fluorescence polarization in growing aggregates, upon excitation with polarized laser light.19 It was possible with this technique to image with high sensitivity the formation of aggregates in an -synuclein-YFP model of aggregation using a confocal microscopy setup with polarization-resolved fluorescence detection.16 The fluorescence lifetime is also a sensitive reporter on the local environment of a fluorophore.20 For example, it was shown that the fluorescence lifetime from appropriate fluorophore labels covalently attached to environmentally responsive polymers informs on the folding kinetics of the latter in live cells.21,22 Notably, it was recently discovered that aggregation into -sheet-rich amyloid fibrils is similarly accompanied by dramatic changes in the fluorescence lifetime. However, here the mechanism differs, and the phenomenon appears to be caused by energy transfer as the molecules become more highly confined in the aggregating protein structures; as a result, the fluorescence lifetime of appropriately selected reporter fluorophores decreases upon fibril formation and this can be used as an in vivo tool to track the aggregation kinetics with microscopy.23 We have used this technique to report, for the first time, on the aggregation of -synuclein in a live Caenorhabditis elegans (C. elegans) model of protein aggregation, see Fig. 3, and were able to correlate the appearance of aggregate species with the emergence of a toxic phenotype.24 Similarly, in a recent study, we have used this technique to track the structural “fate” of amyloid-beta protein ingested in neuronal cell lines and found that the compartmentalization of into small acidic vesicles induces aggregation of the protein, a finding that is significant in context of the disease, providing potential targets for therapeutic strategies.26 Fig. 3Imaging the aggregation state of -synuclein in a live C. elegans model of -synuclein aggregation. (a) Example images of protein expressed in neuronal cells in the live worms. The panel on the left shows a bright-field image of the nematode, on which the fluorescence image of the labeled amyloid is superimposed in green. The right panel shows the corresponding fluorescence lifetimes determined by TCSPC confocal microscopy. (b) Fluorescence lifetime as a function of age for live worm expressing different protein constructs. Black bars indicate -synuclein-YFP; white bars indicate YFP controls; and gray bars indicate Q40-YFP worms. Clearly, a monotonic decrease in the lifetime is observed with age for the worms expressing -synuclein-YFP, indicating increasing progress of aggregation. The observed drop in lifetime has been explained in terms of nonresonant energy transfer processes to absorbing states in the emerging amyloid scaffold,24 but recent work suggests that fluorescence self-quenching also plays an important role in this process.25 The monotonous decrease of the observed lifetimes reflect a mixture of low lifetimes of increasingly aggregated protein that emerges in the aging worm and larger lifetimes of newly synthesized protein that is continuously produced in the nematode. For the strongly aggregating Q40-YFP, the corresponding drop in lifetime is even more rapid than for -synuclein. (c) Increasing aggregation of -synculein leads to an emerging toxic phenotype: From day 8, worms exhibit increasing loss of movement, concurrent with the appearance of small toxic oligomers as verified by TCSPC imaging. Figure reprint with permission from Ref. 24, copyright 2011 Wiley. 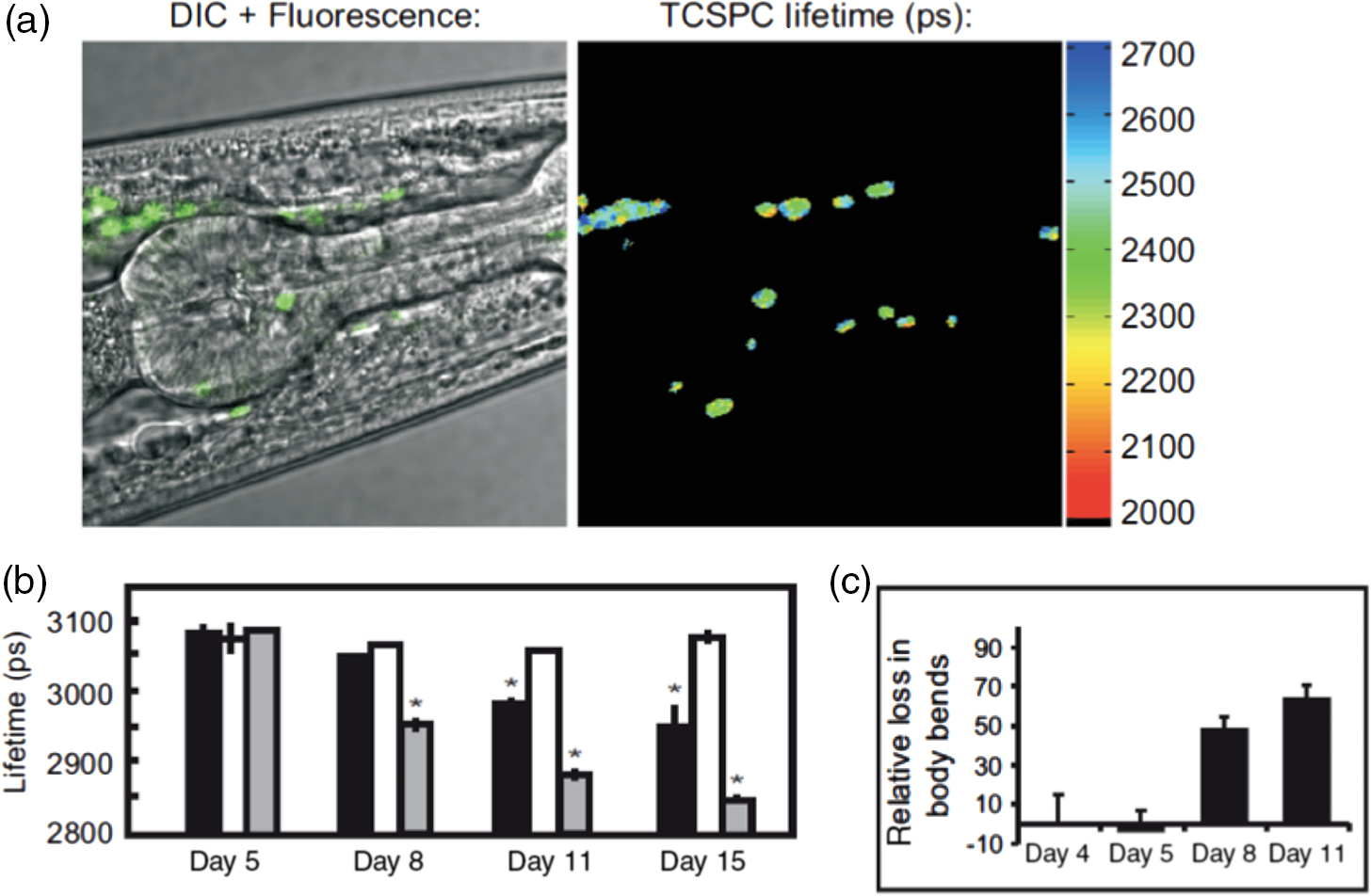 4.Optical Superresolution Imaging of Amyloid FormationWhile multiparameter optical microscopy offers powerful glimpses into the kinetics of aggregation, its optical resolution is limited by diffraction, and it is thus not capable of offering morphological information on the formed species, or to verify the colocalization with subcellular targets, e.g., vesicles or mitochondria, with good enough spatial resolution. The advent of optical superresolution microscopy techniques has led to a paradigm shift in this field. The first application of single-molecule superresolution imaging techniques for the visualization of amyloids was by Duim et al,27 where blink microscopy was used to record fibrils of polyQ protein, a model for Huntington’s disease. Using the technique of direct stochastic optical reconstruction microscopy (dSTORM),28 we were able to image in vitro and in cells the morphology of fibrils with a resolution better than 20 nm.29 The latter study highlighted the influence of the cellular environment on aggregation kinetics and species morphology, differing markedly to what was observed for comparable conditions in vitro, in the test tube. In contrast, fibrillar polyQ species appeared to be more similar between the corresponding in vitro and in cellulo cases, as observed in the study by Sahl et al.30 Similarly, the aggregation of human lysozyme, which occurs in a certain case of hereditary amyloidosis linked to specific lysozyme mutations, was observed both in vitro and in cells using dSTORM and showed similar morphological features.31 In general, such insights are offered now by superresolution imaging methods as their spatial resolution approaches that of AFM or SEM techniques, while offering the advantages of specificity and sensitivity that come with all-optical techniques. In Fig. 4, we show how dSTORM permits the tracking of fibril growth at the single-molecule level, offering information on the self-assembly process, which is not accessible from bulk measurements.32 The images correspond to the “seeded growth” assay of -synuclein as discussed in Sec. 2, for which bulk fluorescence data are shown in Fig. 2. Fig. 4-synuclein elongation assay performed with dSTORM superresolution microscopy. The assay corresponds to the same conditions as for the bulk measurements shown in Fig. 2. Small fibril seed species, shown in green, are incubated in solution containing monomeric -synuclein, shown in red. -synuclein seeds were covalently labeled with Alexa Fluor® 568 and monomer with Alexa Fluor® 647 dyes, respectively. The images show the time-sequenced growth of individual -synuclein fibrils. (a–h) Clearly, growth takes place from both ends of the seed fibril, extending to several micrometers in length after 24 h. The last image (i) shows a conventional fluorescence microscopy image, blurred by optical diffraction. Adapted with permission from Ref. 32, copyright 2014 American Chemical Society. 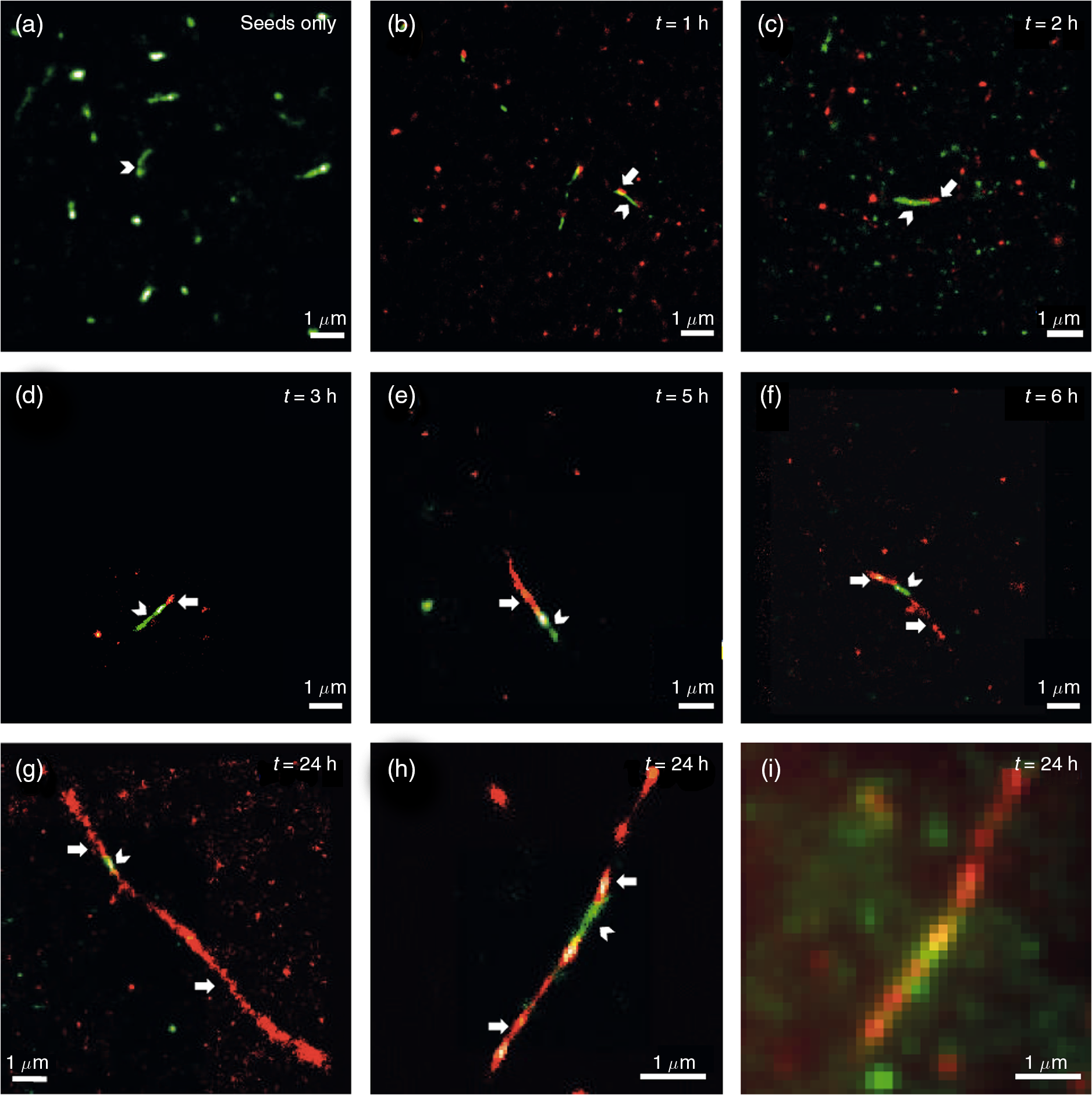 Clearly, growth is seen for individual molecular species. Several insights were obtained from this study that could not have been verified otherwise. First, fibril growth clearly proceeds from both ends of the fibril seeds, a hypothesis, that had been the subject of considerable debate for lack of conclusive evidence. Second, it was possible to show that the growth rates of individual fibrils varied by orders of magnitude; while some fibrils grew hardly at all, others elongated extensively over the same aggregation period.32 The experiment dismissed both random diffusion or the so-called “stop-and-go”’ models of monomer addition, and instead points to the likely existence of several “misfolding states,” for which aggregation can proceed, but which feature individually differing growth rates. It is possible then with such techniques to verify, even in cells,33 whether certain mutants of amyloids “cross-seed” other species, e.g., whether certain toxic species can “force” healthy species to coaggregate. The latter is a variant of the “prion-like propagation” model of amyloid spreading in disease, recently confirmed for -synuclein in neurons.34 This idea was explored for amyloid beta, the protein at the heart of AD, in Ref. 35. In this study, soluble brain and cerbrospinal fluid (CSF) samples from patients with AD were diluted and injected into the hippocampal tissue of young, amyloid precursor protein transgenic mice. When brain tissue from mice thus treated was analyzed 8 months later, it was shown, using superresolution imaging and other techniques, that the capacity to seed the aggregation process is much higher for brain tissue than for CSF, even when the former is massively diluted compared to the latter. An example of a relevant superresolution experiment, performed in vitro, is shown in Fig. 5. Here, extracts from brain samples [Figs. 5(a) and 5(b)] and CSF [Figs. 5(c) and 5(d)] from Alzheimer patients were fluorescently labeled (red) and incubated with differentially labeled monomer (green) in vitro and the aggregation of the latter tracked in time [Figs. 5(e) and 5(f)]. The capacity of brain samples to seed monomer aggregation was found to be much enhanced over that of CSF-induced aggregation. Fig. 5AD CSF reveals limited seeding activity in a single-molecule resolution assay in vitro compared to brain tissue. (a–d) Representative superresolved (a, and c) and diffraction-limited fluorescence (b, and d) images of HF647-labeled amyloid--40 peptide fibrils that were seeded for 72 h with the soluble AD brain fraction (a and b) and AD CSF (c, and d). Scale bar: . (E and F) Higher magnification (four examples) of superresolution images of soluble AD brain fraction-induced aggregates (e) and the AD CSF-induced aggregates (f) (in green, antibody CN3 against human amyloid- peptide; in red, HF647-labeled amyloid--40 peptide). Scale bar: . (g) Measurement of the amyloid--immunopositive particles (labeled green in e and f) revealed smaller amyloid--positive particles in the CSF compared to the amyloid--positive particles in the supernatant (SN) fraction. CN3 does not easily recognize the elongated HF647-labeled amyloid--40 peptide, presumably because of steric hindrance. (h) Mean size of the amyloid--labeled and elongated particles (labeled red in e and f). As a control, amyloid--40 peptide only was used and this measurement was taken as baseline. Consistent with the images shown in a–d, the SN-induced amyloid- aggregates were at least threefold larger compared to the CSF-induced amyloid- aggregates. Five to six randomly chosen areas (as shown in a–d) were analyzed per sample: two SN fractions, and two AD CSF specimens were analyzed. Adapted with permission from Ref. 35, copyright 2014 Oxford University Press. 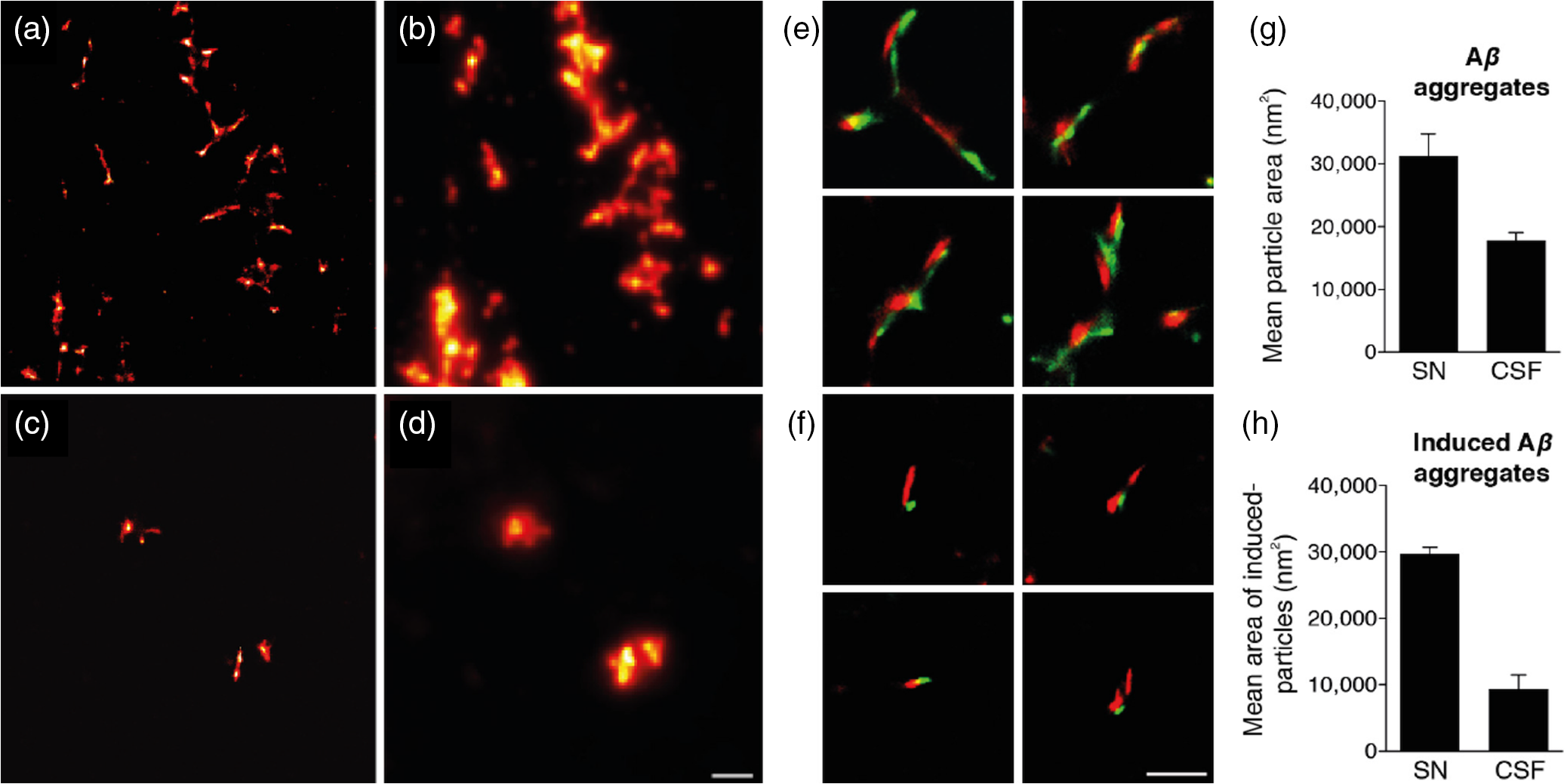 In another study led by Michel et al,36 we used both multiparameter and superresolution imaging to verify how the protein tau propagates in a cell model of tau aggregation. In this study, a short fragment of the human form of the protein tau, K18Tau, covalently labeled with a synthetic fluorophore, was used as a monomeric solution to incubate neuron-like cells (see Fig. 6). Confocal time-correlated single photon counting (TCSPC) microscopy reveals that the protein is readily taken up by cells and is compartmentalized in endocytotic vesicles, small acidic compartments that quite apparently enhance the aggregation rate of the ingested protein. The fluorescence lifetime of the reporter fluorophore drops dramatically upon ingestion into the cells indicating a strong degree of tau aggregation is taking place. This led to the proposal that acidic conditions favor tau aggregation, which was verified in vitro, again via multiparametric imaging (see panels b and c in Fig. 6). Indeed, this finding suggests that uptake of tau via endocytosis is potentially a detrimental process; acidification and crowding upon ingestion into transport vesicles appear to provide conditions by which tau can aggregate. The last panel in Fig. 6(d), shows a two-color superresolution image of fibrillar species found in the extracellular medium, after extended periods of K18Tau incubation of the cells. Intriguingly, the cell expels fibrillar aggregates that contain both the ingested K18Tau, shown in red, and endogenous wild-type tau, already present in the cell, shown in green. The image proves beyond doubt that the extracellular K18Tau, which aggregates on ingestion into neuron-like cells, acts as a template, which “seeds” the coaggregation of endogenous, “healthy” tau. This has major pathological implications, as the model suggests that ingested tau could potentially become toxic and lead to the formation of aggregation nuclei, from which coaggregation of healthy protein species can proceed and propagate from cell to cell. The study furthermore suggests that the mere presence of tau in the extracellular space, e.g., following a brain trauma that causes local cell death and thus release of tau, can have potentially devastating effects; subsequent ingestion by adjacent, healthy cells triggers its aggregation and the consecutive recruitment and coaggregation of endogenous tau, starting a vicious circle, which mimics the disease cascade that may go on in humans. More generally, the study complements our other study on uptake and aggregation26 in suggesting that vesicular compartmentalization and acidification may be quite a general principle by which amyloid aggregation may proceed in cells. Fig. 6Uptake of monomeric tau protein into neuron-like cells causes its aggregation and subsequent recruitment of endogenous tau, normally present in the cell. (a i) Lifetime image of K18Tau, covalently labeled with Alexa Fluor® 488 fluorophore, which does not aggregate in the extracellular space. (a ii) Confocal fluorescence intensity image displaying the distribution of K18Tau inside the cell upon ingestion. Independent experiments confirm compartmentalization in acidic, endocytotic vesicles (not shown). (a iii) TCSPC-fluorescence lifetime images of the ingested protein suggests that significant aggregation has taken place upon protein ingestion. Compared to the incubating solution, shown in a i, the vesicular species show significantly reduced lifetimes, a iii, down by ca. 500 ps. (b) In vitro confirmation by TCSPC to show that low pH leads to aggregation of monomeric tau. (b i) K18Tau at pH 7.4 and (b ii) tau aggregate forming at pH 4.7. (c) Average fluorescent lifetimes of monomers and aggregates shown in b. (d) Incubation of cells with K18Tau leads to the release of fibrillar aggregates, containing both endogenous, intracellular tau, green, coaggregating with the extracellular K18Tau, which was ingested by the cell, shown in red. The two-color dSTORM superresolution experiment confirms a prion-like propagation of misfolded tau species in this cell model, which may be highly relevant to the human form of AD. This figure was originally published in Ref. 36, © the American Society for Biochemistry and Molecular Biology, reproduced here with permission.  5.Conclusions and Future OutlookThis short review has put in focus recent developments in optical imaging that permit us to gain information, at the nanoscale, of the self-assembly process of neurotoxic protein species. Functional imaging techniques, so-called multiparameter imaging techniques, use spectroscopic signature changes to inform on the protein aggregation process. On the other hand, optical superresolution techniques reveal greater spatial detail than has been hitherto possible.30 In combination, these techniques are greatly empowering ways to perform research into neurodegenerative diseases and to probe their molecular origins. A myriad of unresolved questions is now open to “illumination” by these tools, raising hopes to find a cure for these devastating illnesses. An extremely promising technique for studies of amyloid growth kinetics to rival dSTORM is total internal reflection fluorescence microscopy with structured illumination microscopy (TIRF-SIM).37 The technique is capable of providing a spatial resolution in reconstructed images of (see Fig. 7) and has several advantages over dSTORM in the present context.38 It provides excellent image contrast and rejects bulk fluorescence that hampers standard implementations of SIM. Furthermore, it permits dynamic imaging, with frame rates easily possible. The method does not require the use of photoswitchable fluorophores or special buffers and is thus ideally suited for kinetic time-lapse imaging, even in live cells. For screening of small molecule compounds interacting with aggregating proteins and studies of the transmission of toxic species from cell to cell, this may well prove to be the method of choice in the future. Fig. 7TIRF-SIM is capable of simultaneous imaging in multiple colors with sub-100-nm resolution. Panels (a) and (b) demonstrate the resolution enhancement of the method on 100-nm beads labeled with multiple colors and excited at 488 and 640 nm wavelength illumination, respectively. The images are divided across the diagonal to show the improvement obtained in going from wide-field imaging (top left part) to TIRF-SIM (bottom right part). (c) A full width at half maximum fit to bead cross sections for a population of beads demonstrating the excellent resolution achievable with the method, and (d) beta amyloid fibrils labeled with 10% rhodamine derivative (488-nm resolution). Scale bars: . Images reprinted from Ref. 37, with permission. 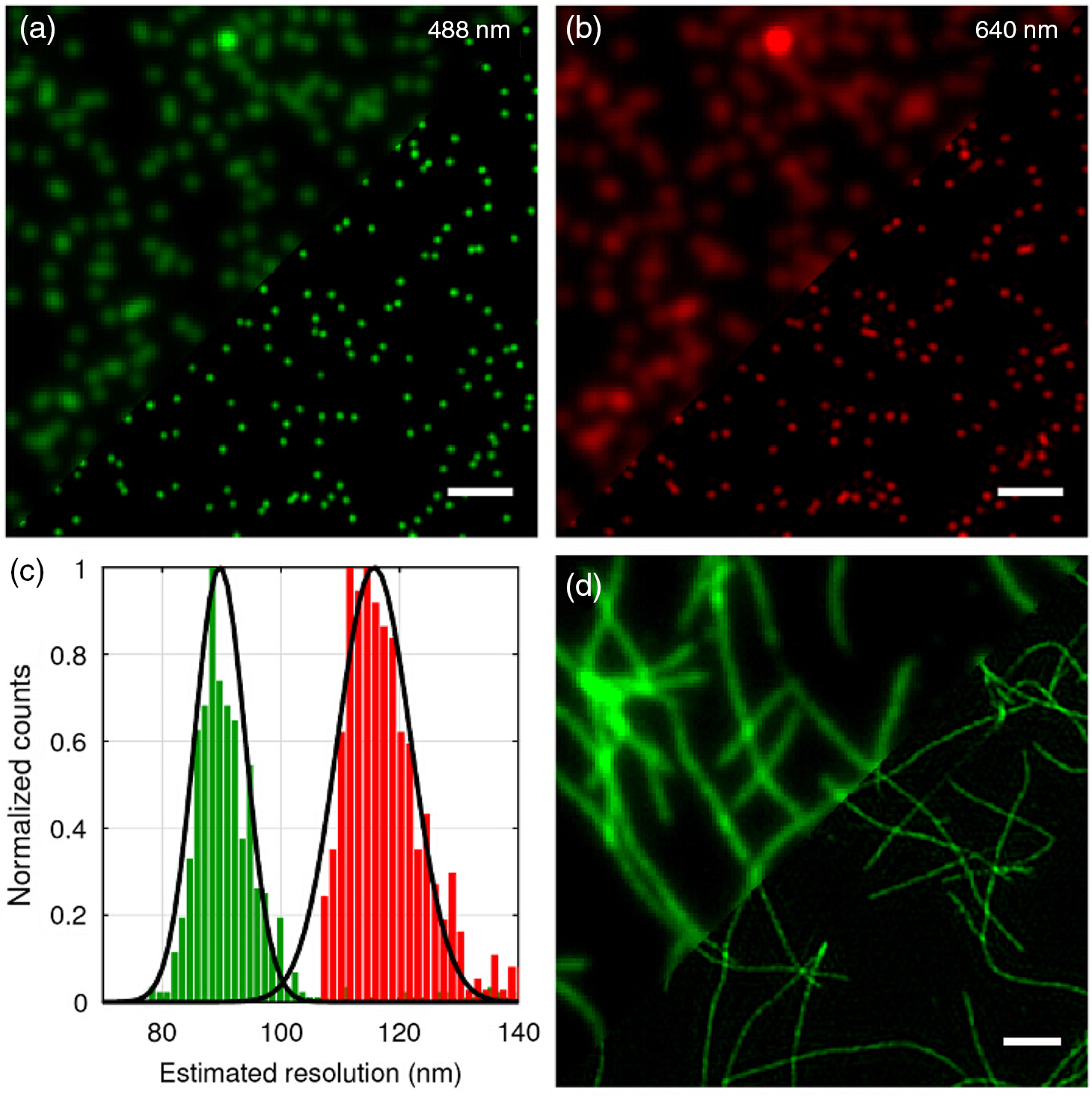 AcknowledgmentsWe wish to thank our many collaborators for their intellectual and practical input to this work, in particular Dr. Claire Michel, who also prepared Fig. 1., Dr. Dora Pinotsi, Dr. Eric Rees, Dr. Miklos Erdelyi, Dr. Amberley Stephens, Dr. Pierre Mahou, Nathan Curry, Florian Ströhl, and Laurie Young. We also thank members of the Cambridge Neurodegenerative Disease Consortium, in particular Professor Chris Dobson, Professor E. E. Mandelkow, Professor P. St. G. Hyslop, and Dr. Seema Quamar. We furthermore wish to thank Professor Markus Sauer and Dr. S. van de Linde for their contributions. This work was funded by grants from the Wellcome Trust, the Medical Research Council UK, the Alzheimer Research UK Trust, the Engineering and Physical Sciences Research Council UK, the Biotechnology and Biological Sciences Research Council, and the Swiss National Science Foundation. ReferencesF. Bemporad and F. Chiti,
“Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships,”
Chem. Biol., 19
(3), 315
–327
(2012). http://dx.doi.org/10.1016/j.chembiol.2012.02.003 CBOLE2 1074-5521 Google Scholar
D. M. Walsh and D. J. Selkoe,
“A beta oligomers—a decade of discovery,”
J. Neurochem., 101
(5), 1172
–1184
(2007). http://dx.doi.org/10.1111/jnc.2007.101.issue-5 JONRA9 0022-3042 Google Scholar
F. Chiti and C. M. Dobson,
“Protein misfolding, functional amyloid, and human disease,”
Annu. Rev. Biochem., 75 333
–366
(2006). http://dx.doi.org/10.1146/annurev.biochem.75.101304.123901 ARBOAW 0066-4154 Google Scholar
S. E. Radford and J. S. Weissman,
“Special issue: the molecular and cellular mechanisms of amyloidosis,”
J. Mol. Biol., 421
(2–3), 139
–141
(2012). http://dx.doi.org/10.1016/j.jmb.2012.05.042 JMOBAK 0022-2836 Google Scholar
Bio-Nanoimaging, 1st ed.Academic Press, Cambridge, Massachusetts
(2014). Google Scholar
E. Cerf et al.,
“Antiparallel beta-sheet: a signature structure of the oligomeric amyloid beta-peptide,”
Biochem. J., 421
(3), 415
–423
(2009). http://dx.doi.org/10.1042/BJ20090379 Google Scholar
K. A. Oberg, J.-M. Ruysschaert and E. Goormaghtigh,
“The optimization of protein secondary structure determination with infrared and circular dichroism spectra,”
Eur. J. Biochem., 271
(14), 2937
–2948
(2004). http://dx.doi.org/10.1111/(ISSN)1432-1033 Google Scholar
C. W. Bertoncini and M. S. Celej,
“Small molecule fluorescent probes for the detection of amyloid self-assembly in vitro and in vivo,”
Curr. Protein Pept. Sci., 12
(3), 205
–220
(2011). http://dx.doi.org/10.2174/138920311795860151 Google Scholar
S. I. A. Cohen et al.,
“From macroscopic measurements to microscopic mechanisms of protein aggregation,”
J. Mol. Biol., 421
(2–3), 160
–171
(2012). http://dx.doi.org/10.1016/j.jmb.2012.02.031 JMOBAK 0022-2836 Google Scholar
F. T. S. Chan et al.,
“Structure-specific intrinsic fluorescence of protein amyloids used to study their kinetics of aggregation,”
Bio-Nanoimaging Protein Misfolding Aggregation, 147
–155 Academic Press, Cambridge, Massachusetts
(2014). Google Scholar
F. T. S. Chan et al.,
“Protein amyloids develop an intrinsic fluorescence signature during aggregation,”
Analyst, 138
(7), 2156
–2162
(2013). http://dx.doi.org/10.1039/c3an36798c ANALAO 0003-2654 Google Scholar
D. Pinotsi et al.,
“A label-free, quantitative assay of amyloid fibril growth based on intrinsic fluorescence,”
ChemBioChem, 14
(7), 846
–850
(2013). http://dx.doi.org/10.1002/cbic.201300103 Google Scholar
D. Pinotsi et al.,
“Proton transfer and structure-specific fluorescence in hydrogen bond-rich protein structures,”
J. Am. Chem. Soc., 138
(9), 3046
–3057
(2016). http://dx.doi.org/10.1021/jacs.5b11012 JACSAT 0002-7863 Google Scholar
G. Gaudriault and J. P. Vincent,
“Selective labeling of alpha- or epsilon-amino groups in peptides by the Bolton–Hunter reagent,”
Peptides, 13
(6), 1187
–1192 http://dx.doi.org/10.1016/0196-9781(92)90027-Z Google Scholar
S. Barghorn and E. Mandelkow,
“Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments,”
Biochemistry, 41
(50), 14885
–14896
(2002). http://dx.doi.org/10.1021/bi026469j Google Scholar
T. J. van Ham et al.,
“Towards multiparametric fluorescent imaging of amyloid formation: studies of a YFP model of alpha-synuclein aggregation,”
J. Mol. Biol., 395
(3), 627
–642
(2010). http://dx.doi.org/10.1016/j.jmb.2009.10.066 JMOBAK 0022-2836 Google Scholar
M. Meyer-Luehmann et al.,
“Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease,”
Nature, 451
(7179), 720
–724
(2008). http://dx.doi.org/10.1038/nature06616 Google Scholar
T. Murakami et al.,
“ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism,”
Hum. Mol. Genet., 21
(1), 1
–9
(2012). http://dx.doi.org/10.1093/hmg/ddr417 HMGEE5 0964-6906 Google Scholar
F. T. S. Chan, C. F. Kaminski and G. S. Kaminski Schierle,
“HomoFRET fluorescence anisotropy imaging as a tool to study molecular self-assembly in live cells,”
ChemPhysChem, 12
(3), 500
–509
(2011). http://dx.doi.org/10.1002/cphc.201000833 Google Scholar
J. W. Borst and A. J. W. G. Visser,
“Fluorescence lifetime imaging microscopy in life sciences,”
Meas. Sci. Technol., 21 102002
(2010). http://dx.doi.org/10.1088/0957-0233/21/10/102002 MSTCEP 0957-0233 Google Scholar
X. Dai et al.,
“A spectroscopic study of the self-association and inter-molecular aggregation behaviour of pH-responsive poly(l-lysine iso-phthalamide),”
Polymer, 47
(8), 2689
–2698
(2006). http://dx.doi.org/10.1016/j.polymer.2006.02.059 POLMAG 0032-3861 Google Scholar
X. Dai et al.,
“Fluorescence intensity and lifetime imaging of free and micellar-encapsulated doxorubicin in living cells,”
Nanomed. Nanotechnol. Biol. Med., 4
(1), 49
–56
(2008). http://dx.doi.org/10.1016/j.nano.2007.12.002 Google Scholar
G. S. K. Schierle, M. Sauer, C. F. Kaminski,
“Probing amyloid aggregation and morphology in situ by multiparameter imaging and super-resolution fluorescence microscopy,”
Bio-Nanoimaging Protein Misfolding Aggregation, 105
–120 Academic Press, Cambridge, Massachusetts
(2014). Google Scholar
G. S. Kaminski Schierle et al.,
“A FRET sensor for non-invasive imaging of amyloid formation in vivo,”
ChemPhysChem, 12
(3), 673
–680
(2011). http://dx.doi.org/10.1002/cphc.201000996 Google Scholar
W. Chan et al., Google Scholar
E. K. Esbjörner et al.,
“Direct observations of amyloid self-assembly in live cells provide insights into differences in the kinetics of and aggregation,”
Chem. Biol.,
(2014). Google Scholar
W. C. Duim et al.,
“Sub-diffraction imaging of huntingtin protein aggregates by fluorescence blink-microscopy and atomic force microscopy,”
ChemPhysChem, 12 2387
–2390
(2011). http://dx.doi.org/10.1002/cphc.v12.13 CPCHFT 1439-4235 Google Scholar
M. Heilemann,
“Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes,”
Angew. Chem. Int. Ed., 47
(33), 6172
–6176
(2008). http://dx.doi.org/10.1002/anie.200802376 Google Scholar
G. S. Kaminski Schierle et al.,
“In situ measurements of the formation and morphology of intracellular -amyloid fibrils by super-resolution fluorescence imaging,”
J. Am. Chem. Soc., 133
(33), 12902
–12905
(2011). http://dx.doi.org/10.1021/ja201651w JACSAT 0002-7863 Google Scholar
S. J. Sahl et al.,
“Cellular inclusion bodies of mutant huntingtin exon 1 obscure small fibrillar aggregate species,”
Sci. Rep., 2 895
(2012). http://dx.doi.org/10.1038/srep00895 Google Scholar
M. Ahn et al.,
“Analysis of the native structure, stability and aggregation of biotinylated human lysozyme,”
PLoS One, 7
(11), e50192
(2012). http://dx.doi.org/10.1371/journal.pone.0050192 Google Scholar
D. Pinotsi et al.,
“Direct observation of heterogeneous amyloid fibril growth kinetics via two-color super-resolution microscopy,”
Nano Lett., 14
(1), 339
–345
(2013). http://dx.doi.org/10.1021/nl4041093 NALEFD 1530-6984 Google Scholar
M. J. Roberti et al.,
“Imaging nanometer-sized -synuclein aggregates by superresolution fluorescence localization microscopy,”
Biophys. J., 102
(7), 1598
–1607
(2012). http://dx.doi.org/10.1016/j.bpj.2012.03.010 BIOJAU 0006-3495 Google Scholar
D. Pinotsi et al.,
“Nanoscopic insights into seeding mechanisms and toxicity of -synuclein species in neurons,”
Proc. Natl. Acad. Sci. U.S.A., 113
(14), 3815
–3819
(2016). http://dx.doi.org/10.1073/pnas.1516546113 Google Scholar
S. K. Fritschi et al.,
“Highly potent soluble amyloid- seeds in human Alzheimer brain but not cerebrospinal fluid,”
Brain, 137
(11), 2909
–2915
(2014). http://dx.doi.org/10.1093/brain/awu255 BRAIAK 0006-8950 Google Scholar
C. H. Michel et al.,
“Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology,”
J. Biol. Chem., 289
(2), 956
–967
(2014). http://dx.doi.org/10.1074/jbc.M113.515445 Google Scholar
L. J. Young, F. Ströhl and C. F. Kaminski,
“A guide to structured illumination TIRF microscopy at high speed with multiple colours,”
J. Visual. Exp., 21
(6), 732
–742
(2016). http://dx.doi.org/10.3791/53988 Google Scholar
F. Ströhl and C. F. Kaminski,
“Frontiers in structured illumination microscopy,”
Optica, 3
(6), 667
–677
(2016). http://dx.doi.org/10.1364/OPTICA.3.000667 Google Scholar
BiographyClemens F. Kaminski is a professor of chemical physics and the Head of the Laser Analytics Group at the University of Cambridge. His group develops modern microscopy techniques and, in close collaboration with his wife Gabi, he studies aspects of neurodegeneration. He is a Fellow of the Optical Society of America and Director of the Engineering and Physical Sciences Research Council UK Centre for Doctoral Training in Sensor Technologies and Applications. Gabriele S. Kaminski Schierle is an associate professor of molecular biotechnology at the University of Cambridge, where she heads the Molecular Neuroscience Group. Her primary interest is in gaining a molecular-level understanding of protein misfolding phenomena associated with neurodegeneration. She works with Clemens to develop and apply superresolution and functional imaging methods in cellular and organism models of disease. |



