
|
|
1.Introduction: Quantitative NeurobiologyThe transmission of signals across chemical synapses entails a precise sequence of events. Neurotransmitters released from the presynaptic terminal into the synaptic cleft activate neurotransmitter receptors in the postsynaptic membrane, thus giving rise to ion fluxes and triggering complex signaling processes in the receiving neuron (Fig. 1). Each of these steps is strongly dependent on the numbers of molecules that are implicated. This is because the rates of molecular interactions are determined by the effective concentration of the reactants. Molecule numbers at individual synapses are relatively small and highly variable, meaning that the strength of synaptic transmission is equally variable and shaped by stochastic events.1 For example, the absolute number of glutamate receptors at excitatory synapses may vary considerably between zero and more than 100, depending on the activity state of the synapse (reviewed in Ref. 2). Neurotransmitter receptors accumulate in the synaptic membrane through molecular interactions with specific scaffold proteins in the postsynaptic density (PSD). With up to several hundred copies, the scaffold proteins generally outnumber the bound receptor complexes. This excess of binding sites results from a dynamic equilibrium between receptors and scaffold proteins according to their binding properties, where changes in affinity can tune receptor numbers and induce synaptic plasticity. Correlated changes at presynaptic and postsynaptic terminals indicate that the relative amounts of some synaptic components are closely coregulated,3 whereas homeostatic mechanisms show that protein levels can be maintained in absolute terms also.4 In short, protein numbers are essential to formulate reaction processes among interacting molecules at synapses. Fig. 1Where numbers count: quantitative parameters of synaptic components. Synaptic transmission ultimately depends on the activity of postsynaptic receptor complexes. The release of neurotransmitters from the presynaptic terminal controls the activation of these receptors, whereas scaffold proteins in the PSD regulate their number and subsynaptic distribution. Precise knowledge of the relevant parameters can help model the underlying molecular mechanisms on a quantitative level. 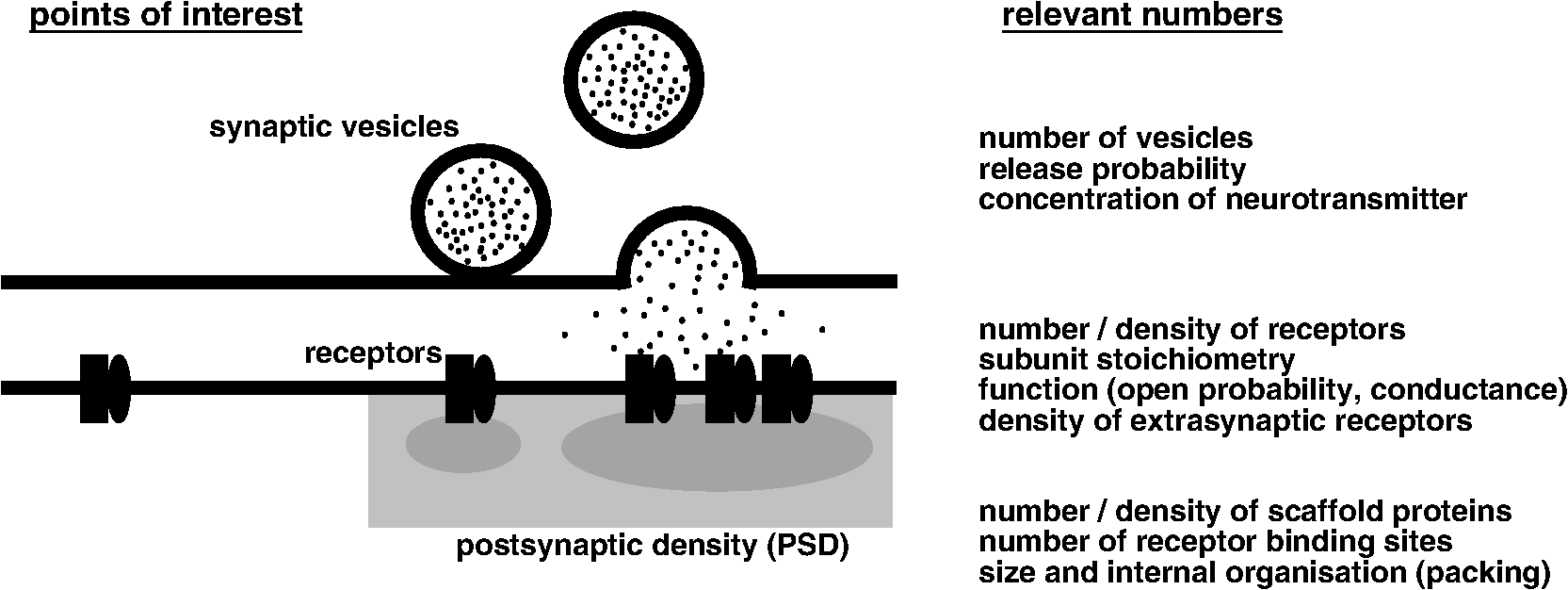 Numbers are equally relevant at the level of individual functional protein complexes. Ligand-gated ion channels are stoichiometric assemblies of several subunits that confer specific properties to receptor subtypes. N-methyl-d-aspartate receptors (NMDARs), for instance, are heterotetramers composed of two GluN1 subunits along with two GluN2A-D and/or GluN3A/B subunits.5 GluN2A and GluN2C-containing receptors display very different decay kinetics and blockage by ions and are deployed in a cell-type specific manner.6 The same is true for other ionotropic channels such as the pentameric -aminobutyric acid A receptor (), whose function, pharmacology and cellular localization depends on the combination of 19 different subunits. For example, containing subunit preferentially localize at extrasynaptic radixin clusters,7 thereby contributing predominantly to tonic inhibition.8 Other subunits target to inhibitory synapses, where they compete with glycine receptors (GlyRs) for binding sites in the postsynaptic scaffold.9 The importance of quantitative neurobiology thus lies in its capacity to explain the function and behavior of proteins and to build realistic models of complex processes. For decades, tools have been developed to determine absolute numbers and stoichiometries, including quantitative molecular biology and proteomics, electrophysiological recordings and electron microscopy (EM), as well as strategies based on calibrated fluorescence imaging. Even though all of these techniques have specific strengths, single molecule localization microscopy (SMLM) has a unique potential to gain quantitative information about several molecule species at behaving synapses at once. Single molecule counting can, therefore, provide new insights into the functioning of synapses that are not easily obtained with earlier methods. 2.Absolute and Relative Proteomic Quantification of Synaptic ComponentsIn 1982, Betz and colleagues10 purified GlyRs from rat spinal cord tissue. Sodium-dodecyl sulfate polyacrylamide gel electrophoresis and silver staining of the isolated receptors identified bands of 48, 58, and 93 kDa that were thought to represent different subunits of the GlyR complex. It was found later that they correspond to the GlyR subunits and and the inhibitory synaptic scaffold protein gephyrin, respectively.11 Chemical crosslinking finally revealed that GlyRs are in fact heteropentameric complexes composed of and subunits with a putative stoichiometry.12 These studies exemplify the usefulness of classical molecular biological techniques for bulk protein quantification. X-ray crystallography has since demonstrated that the pentameric structure is common to other ligand-gated ion channels such as the and nicotinic acetylcholine receptors (nAChRs) and has important implications for the pharmacological profile of this receptor family.13 On a larger scale, a variety of proteomic strategies have been developed for the quantification of neuronal proteins in complex samples.14 These methods generally require the purification of enriched fractions from intact tissue (e.g., PSDs, synaptosomes, membrane fractions, or synaptic vesicles), followed by enzymatic cleavage of the proteins, the fractionation of peptide samples and mass spectrometric (MS) analysis. Accurate profiling of peptide levels can be done using isobaric tags such as the multiplex isobaric tagging for relative and absolute quantification (iTRAQ) reagents that can be used for differential labeling of up to eight samples. The MS spectra of the combined samples reveal not only the peptide sequence but also the relative amounts of the peptides in the different samples. Making use of this approach, activity-dependent changes of about 900 synaptic proteins were measured at four time points (0, 10, 20, and 60 min) after intraperitoneal injection of pilocarpine.15 This showed that functionally related proteins such as various glutamate receptor subunits and scaffold proteins (e.g., GluA1-3, GluN1/2A/2B, PSD-95, and Shank1) are coregulated, pointing to the existence of core complexes within the PSD whose components act in concert to regulate synaptic activity. For the absolute quantification of proteins using MS, known concentrations of labeled standards that are chemically indistinguishable from the proteins/peptides of interest can be added to the samples and processed simultaneously. As they behave similarly to the analyzed material, their intensities are used to calculate the concentration of the respective component. One such absolute quantification approach (AQUA) has been applied to 32 core proteins at excitatory PSDs from rat forebrain.16 The most abundant proteins were and , accounting for 7.4% and 1.3% (w/w) of the mass of the PSD. The scaffold protein PSD-95 was also highly enriched (0.7%) and exceeded the copy number of glutamate receptor subunits by an order of magnitude. The ratio among different -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) subunits suggested that forebrain synapses express mostly GluA1/GluA2 heteromers and only a minority of GluA2/GluA3. This subunit stoichiometry is strongly region-dependent, as shown in a recent quantitative proteomic characterization of purified AMPAR complexes.17 Protein concentrations can be converted into absolute copy numbers if the total mass of the PSD is known. A study based on quantitative scanning transmission electron microscopy and gel electrophoresis estimated the total mass of the PSD to be 1.1 GDa.18 An average PSD contained copies of PSD-95 that contribute about 2.3% of its mass. If applied to the data of Cheng et al.16, a structure of 1.1 GDa should contain in the order of 100 molecules of PSD-95 (0.7% w/w). The discrepancy between the two studies is probably due to differences in sample preparation, and highlights one of the main drawbacks of quantitative neuroproteomics that is the biochemical purification of protein samples at the loss of cell type and synapse-specificity (Table 1). Nonetheless, the high throughput and versatility make quantitative MS a powerful tool for large-scale protein profiling, including the analysis of alternative splice variants or posttranslational modifications (e.g., Ref. 19). Table 1Comparison of quantification strategies and readouts.
3.Quantification of Functional Receptors at Synapses Using ElectrophysiologyThe strength of synaptic transmission results from the activity of postsynaptic neurotransmitter receptors. It is influenced by several parameters: the concentration of the neurotransmitter in the synaptic cleft, the number of available receptors in the postsynaptic membrane, and their channel properties (open probability, conductance, and kinetics). Hence, ion fluxes can serve as a readout of receptor numbers, provided that the other parameters can be measured somehow. Essentially, the number of receptors activated by a neurotransmitter quantum () can be calculated as the response of a population of receptors (postsynaptic current) divided by the response generated by a single receptor. In some cases, the currents conducted by single receptors can be directly observed through patch-clamp recordings,20,21 as illustrated by the discrete single channel conductance steps of of 14 and 23 pS.22 With a conductance of 140 to 400 pS, inhibitory postsynaptic currents (IPSCs) evoked by single quanta in dentate gyrus granule cells hence involve the activity of a maximal range of 6 to 30 , on the assumption that the extrasynaptic and synaptic receptor populations are the same. When the channel properties prohibit the detection of single channel conductance steps, these can be calculated using nonstationary fluctuation analysis.23 The relationship between currents () and their variance around the mean () is fitted with a parabolic function that gives the single channel current (), the number of available receptors () and their open probability (). Using this approach, Jonas et al.24 have recorded AMPA/kainate receptor currents evoked with 3-mM glutamate in outside-out patches of hippocampal CA3 pyramidal neurons, and determined an elementary channel conductance of 8.5 pS and an open probability of 0.71. If applied to unitary excitatory postsynaptic currents at mossy fiber synapses on CA3 pyramidal neurons (121 to 553 pS), this results in a number of 14 to 65 open receptors per quantal event and 20 to 92 total receptors. Direct comparison between nonstationary fluctuation analysis and observable conductance steps often shows good correspondence. Single channel conductance derived from glycinergic miniature mIPSCs and from outside-out patches of motoneurons in the brain stem was almost identical with channel closings in the decay trace of the patch currents (), implying a homogeneous GlyR population.25 Taking into account the open probability in response to 1-mM glycine, the total numbers of synaptic GlyRs were 27 in neonate and 39 in juvenile rats. However, variations in the decay time course suggested developmental changes as well as possible differences between synaptic and extrasynaptic GlyRs.25 In other words, this quantification strategy relies on certain assumptions, such as the neurotransmitter concentration in the synaptic cleft, the homogeneity of the receptor population, or the electric properties resulting from the morphology of the neuron. It also has to be kept in mind that electrophysiology is restricted to the quantification of active receptors (Table 1). The existence of transiently inactive (desensitized) receptors or differences in receptor occupancy following the release of a single, nonsaturating quantum may lead to significant underestimation of the total number of synaptic receptors.26 To address some of these issues, patch-clamp recordings have been used together with immuno-EM in what is a great combination of techniques that provides at once absolute numbers of receptor subunits at individual synapses and ultrastructural information. 4.Quantification of Receptor Subunits Using Calibrated Immunogold LabelingImmunogold labeling of the subunit in cerebellar stellate cells showed a large variability in particle numbers in serially reconstructed synapses, ranging from 6 to 184 around a mean of 56 gold particles per PSD.26 The mIPSC amplitudes in these neurons had a similar, skewed distribution, indicating that the number of immunogold particles correlates with activity, even though the two types of data had been obtained independently in fixed tissue and acute slices, respectively. Using nonstationary fluctuation analysis of mIPSCs, the authors determined a number of 140 total receptors at an average synapse, resulting in a conversion factor of complexes per gold particle. In this way, the immunogold data could be calibrated in absolute terms. The high spatial resolution of EM makes it possible to relate these numbers to the ultrastructure of the synapse. For instance, the linear relationship between receptor number and PSD area demonstrated that are distributed at a uniform density of about at synapses.26 In principle, the calibration of immunogold labeling can be applied to any other cell type or subcellular compartment. For example, densities at synapses in cerebellar granule cells are about two orders of magnitudes higher than those in the extrasynaptic membrane.27 The synaptic enrichment is dependent on the availability of binding sites and the affinity of receptor-scaffold interactions. The comparison of protein densities in absolute terms may therefore reveal new types of information about the strength of these biochemical interactions within cells. Due to the time-consuming nature of immuno-EM, however, no comprehensive quantifications of different receptor subunits across brain regions have been conducted. 5.Quantification of Synaptic Protein Populations Using Calibrated Fluorescence MicroscopyThe determination of protein levels based on fluorescence microscopy has become a widely used tool in neurobiological research due to its relative simplicity and the possible detection of almost any molecular target. Populations of proteins are usually visualized with specific antibodies coupled with organic fluorophores or by expressing recombinant proteins fused with a fluorescent protein. Relative changes in protein abundance can easily be measured either in fixed or in living cells, and are expressed in arbitrary units of fluorescence intensity. Absolute quantification, however, is less straightforward, since it requires knowledge of the fraction of labeled molecules and of the fluorescence associated with a single fluorophore (Table 1). As a way to convert fluorescence into molecule numbers, the intensity of single GFP fluorophores was used to calibrate beads to which increasing amounts of GFP had been bound.28 The exact amount of protein on the beads was determined by quantitative amino acid analysis, suggesting that at least 85% of GFP molecules were functional (i.e., fluorescent). The proportion of labeled proteins within a cell is often unknown due to the variability of antibody labeling or the overexpression of recombinant proteins. The use of knock-in animals that express fluorescently tagged proteins at endogenous levels offers a solution. This is illustrated by a fluorescence calibration study measuring the absolute numbers and densities of the GABA transporter GAT1 at inhibitory synapses.29 Endogenous GAT1-GFP was visualized with confocal microscopy and calibrated with beads and polyacrylamide gel slices containing known concentrations of GFP. The analysis showed that GAT1 levels differ strongly between synapses, with an average of 1200 copies at presynaptic boutons in hippocampus and 9000 in cerebellar basket cells. Considering the respective size of these boutons, however, the calculated membrane densities were far less variable at about 800 to . This may indicate that the two-dimensional packing of GAT1 is relatively equal despite the large differences in their total amounts. The use of knock-in models to quantify copy numbers may not always be necessary, if the degree of overexpression of a recombinant protein can be assessed. In a detailed quantification of scaffold proteins at excitatory synapses, the level of overexpression was calculated by comparing the immunoreactivity in neurons expressing enhanced (E)GFP-tagged recombinant proteins with those in control neurons.30 Small fluorescent microspheres were used to calibrate the fluorescence intensity of single EGFP molecules. Taking into account the chromophore formation efficiency (65% to 80%), the authors concluded that an average excitatory PSD contains about 300 copies of endogenous proteins of the MAGUK family, 200 of which are PDS-95. In addition, 170 GKAPs, 310 Shank proteins, and 350 copies of the Homer family of scaffold proteins were found. Their relative abundance differed somewhat from that obtained by quantitative proteomic analysis ().16 The most likely cause for this discrepancy is the loss of PSD components during biochemical purification, which may differentially affect scaffold proteins according to their stability in the PSD.31 A major limitation of conventional fluorescence microscopy comes from its low spatial resolution. This is exemplified by the detection of labeled receptors or scaffold proteins in the PSD that can be easily visualized and quantified in relative terms [Fig. 2(a)]. The fluorescence intensity of labeled GlyRs at synaptic gephyrin clusters, for example, is about twofold to threefold higher than in dendrites.32 This apparent enrichment factor is a serious underestimation, due to the low resolution of conventional fluorescence microscopy that magnifies the apparent size of the synaptic domain. According to high resolution EM data, receptors are concentrated to a much greater extent at synapses.27 Hence, the quantification of protein concentrations in small cellular compartments requires not only the conversion of arbitrary fluorescence units into molecule numbers, but also precise spatial information (see Sec. 7). Fig. 2Molecule counting using conventional and single molecule imaging. (a) Conventional fluorescence microscopy of a dissociated hippocampal neuron expressing Dendra2-tagged . (b) Idealized representation of an intensity-time trace displaying photobleaching steps. (c) Histogram of fluorophore counts (black bars) and data fitting with a binomial distribution corresponding to two fluorophores and a probability of detection of 80% (gray). Note that some overcounting may occur in these experiments. The counts have been corrected for missed events where both fluorophores are inactive (). (d) Pointillist image of Dendra2- detections of the neuron shown in (a). Clusters of detections belonging to the same molecule can be spatially resolved in sparse areas (inset). (e) Illustration of a time trace of PALM detections. Bursts of detections originating from different fluorophores can be separated temporally to determine molecule numbers. The number of bursts can be fitted with a binomial distribution to account for the photoactivation efficiency of the fluorophore [as in (c)]. (f) Pulsed photoconversion of subsets of fluorophores in a dense structure. Steps in the decay traces correspond to the bleaching of single fluorophores [as in (b)]. The total number of fluorophores can be calculated as the sum of the peak intensities expressed in absolute numbers. (g) Quantitative analysis of fluorescence decay kinetics. The number of molecules in a dense structure can be calculated as , where is the area under the curve (total intensity of the structure), is the average intensity of individual fluorophores, and is the lifetime of the fluorophores. (h) Pair correlation analysis of the spatial distribution of fluorophores. Deviations from a random distribution (dots) identify clustering due to recurrent fluorophore detections (gray) or the presence of protein complexes (black curve) at characteristic distances. 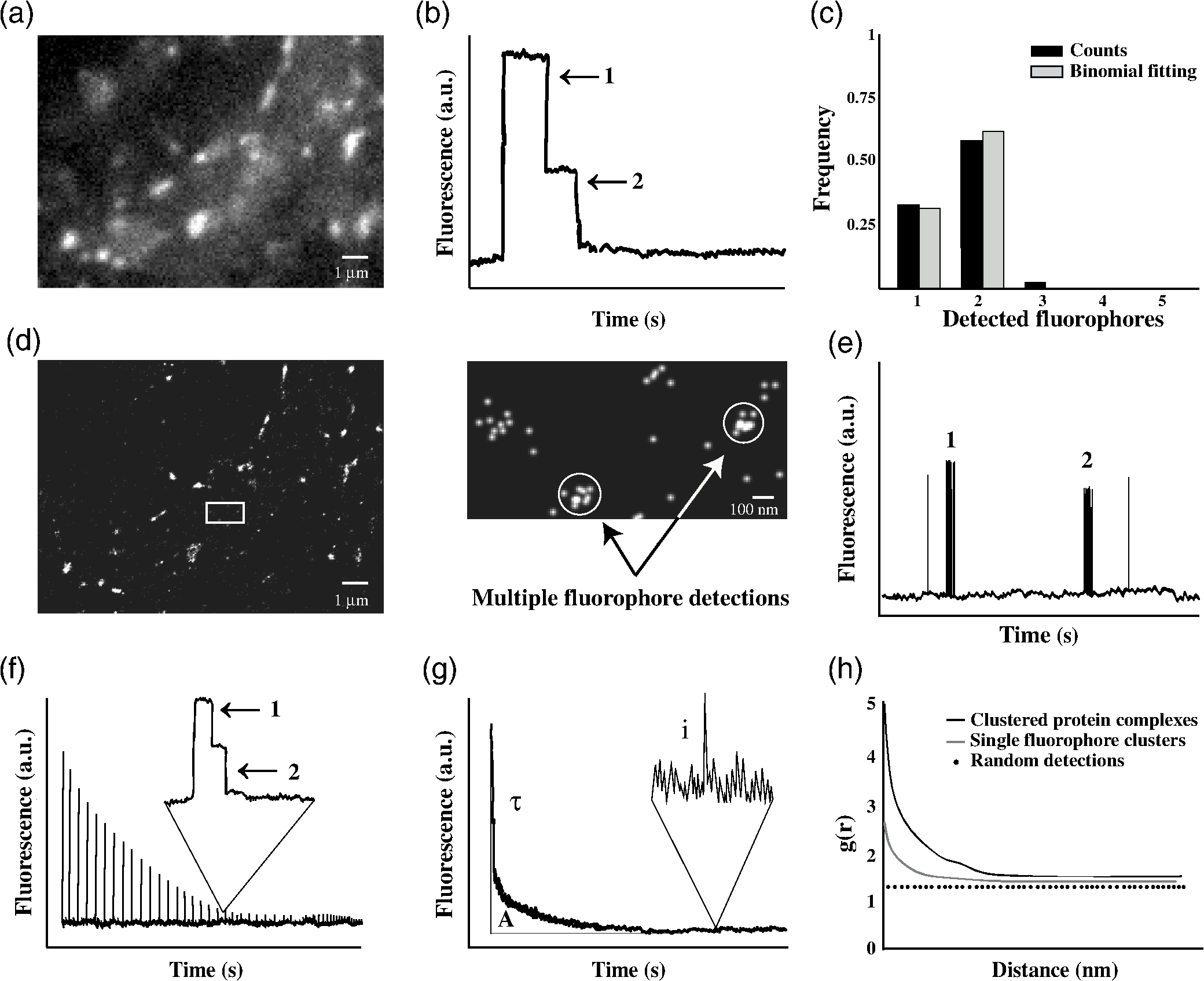 6.Counting of Single Receptor Subunits with Conventional FluorophoresThe use of fluorescence microscopy for the characterization of receptor stoichiometry was first done by counting single bleaching steps of EGFP-tagged NMDARs.33 Tagged receptor subunits were coexpressed with nonfluorescent subunits in Xenopus laevis oocytes. Sparsely distributed receptor complexes on the cell surface were imaged with total internal reflection microscopy until fully bleached. Discrete steps of photobleaching in the decay trace correspond to the number of fluorescent proteins in the receptor complex [Fig. 2(b)]. These experiments confirmed that NMDARs contain two GluN1 subunits per tetramer34 in support of crystallographic data.35,36 A minority of the visualized spots of GluN1-EGFP displayed a single bleaching step due to the existence of nonfluorescent EGFP molecules.33 In other words, since the probability to detect the target proteins is , the presence of two subunits results in a binomial distribution of either one or two observed bleaching steps [Fig. 2(c)]. To calculate the fraction of nonfunctional EGFP, the distribution of counts for a given stoichiometry can be fitted with a binomial distribution, with the probability of detection () as free parameter. In these experiments EGFP fusion proteins had a probability of about 80%, similar to previous studies.30 Zero bleaching steps consequently occur with a frequency of 0.04 if neither of the two subunits is fluorescent, but these events are obviously not recorded. Overcounting ( steps) can happen if more than one protein complex is located within a diffraction-limited spot. The difficulty in quantifying these data thus lies in the few available data points for fitting (bins 1 and 2) and the strong influence of counting errors (discussed in Ref. 37, supporting material). Nonetheless, this method has been widely employed to study heteromeric receptor complexes as it does not require very specialized optical equipment. Dual-color recordings have shown the number of binding sites of AMPARs for auxiliary transmembrane AMPAR regulatory proteins as well as the occupancy of these sites (Ref. 38, see also Ref. 39). The number of bungarotoxin binding sites of nAChRs was determined by counting the bleaching steps of Alexa Fluor 647-conjugated ligand.37 Furthermore, GlyR stoichiometry was assessed using YFP-tagged or subunits coexpressed with unlabelled subunits in Xenopus oocytes.40 The results lend support to a model whereby heteromeric GlyRs are composed of three and two subunits,12,41,42 although an stoichiometry has also been proposed.43,44 This has important consequences for the accumulation of GlyRs at synapses since the subunit mediates the interaction with the scaffold protein gephyrin.9 Taken together, these studies illustrate the impact that the discovery of GFP and its variants has had not only on fluorescence microscopy as such, but also on quantitative imaging. GFP-calibration strategies can be used to study most membrane-associated or intracellular proteins, as there are no general constraints on the class of molecules that can be fluorescently tagged. A serious limitation arises from the low spatial resolution of light microscopy, whereby two structures at a distance of cannot be spatially resolved. This is why the counting of bleaching steps is restricted to sparsely expressed protein complexes. A related source of error lies in the calculation of absolute protein densities in small cellular compartments such as the PSD, which requires both accurate molecule numbers as well as ultrastructural information to resolve the distribution and packing of synaptic proteins. 7.Counting Synaptic Proteins with Single Molecule Localization MicroscopySeveral recent super-resolution imaging techniques bypass the limitations of conventional fluorescence microscopy (reviewed in Ref. 45). Among these, photoactivated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM) hinge on the detection of single photoactivatable or switchable fluorescent proteins or organic dyes.46–48 As these probes change their emission spectrum in a controlled manner (e.g., upon illumination with near–UV light), single molecule data are gathered by temporally resolving individual fluorophores. Their locations can be calculated with a precision in the nanometer range (typically with currently available probes, Table 1). The information belonging to many fluorophores is then combined to obtain a super-resolution pointillist image [Fig. 2(d)]. As an example, the subsynaptic distribution of several pre- and postsynaptic proteins at excitatory synapses (e.g., Piccolo, Bassoon, GluA1, GluN2B, CaMKII, PSD-95, Shank, and Homer) was mapped using multicolor three-dimensional-STORM.49 The data provide striking evidence that receptors and scaffold proteins are organized in laminar fashion and lie at different distances from the synaptic cleft (discussed in Ref. 50). Similarly, the combination of various super-resolution techniques including PALM-based single particle tracking (sptPALM) and STORM disclosed the presence of nanodomains of AMPARs and PSD-95 at hippocampal synapses.51 These studies give insight into the internal organization of synapses that underlies signaling processes on the molecular scale. The recording of discrete numbers of detections makes SMLM an inherently quantitative approach, where the number of detections reflects the number of fluorophores (e.g., Ref. 52). Unfortunately, the ill-defined photophysical properties of fluorophores complicate the use of SMLM for absolute quantification.53 Since a fluorophore may be active for several consecutive frames and its calculated location varies due to stochastic fluctuations, each fluorophore is in fact represented by a small cluster of points in a pointillist image [Fig. 2(d)]. As a consequence, the number of detections that constitute the pointillist image is not equal to the number of molecules present. Instead, bursts of detections must be translated into absolute molecule numbers. The conversion is relatively straightforward when the events are well separated in space and time [Figs. 2(d) and 2(e)]. The stoichiometry of GlyR complexes expressed in Xenopus oocytes was analyzed in this way.54 As for the counting of bleaching steps, the observed burst frequencies were fitted with a binomial distribution to calculate the underlying stoichiometry as well as the photoactivation efficiency of the fluorophores [in analogy to the probability of detection ; Fig. 2(c)]. The data reaffirm the model discussed earlier. What is more, this study compared the behavior of different photoactivatable proteins used for SMLM. About 60% of mEos2, 50% of Dendra2, but only 40% of PA-GFP molecules were successfully photoactivated and detected under the chosen experimental conditions.54 A disadvantage of quantitative PALM consists in the difficulty to count large numbers of proteins in dense compartments,55 as is the case for scaffold proteins at synapses. A combination of methods has allowed us to estimate the absolute number of gephyrin scaffold proteins at inhibitory synapses.56 As a first approach, we analyzed the fluorescence decay traces of small subsets of photoconverted Dendra2-tagged gephyrin to identify bleaching steps. This is ultimately the same strategy as the one described by Ulbrich and Isacoff,33 only that it was applied to subsets of a large population of fluorophores using pulsed photoconversion. We could then translate the peak intensities into absolute numbers, the sum of which represents the total number of scaffold molecules in a given cluster [Fig. 2(f)]. According to our data, inhibitory synapses in cultured spinal cord neurons contained an average of 200 recombinant gephyrin molecules.56 As an independent approach, we measured the decay kinetics of endogenous mRFP-gephyrin clusters in spinal cord neurons from knock-in mice. The total fluorescence intensity of a cluster is given by the area under the decay trace [Fig. 2(g)]. This value was divided by the total unitary fluorescence, calculated as the product of the intensity of single blinking events () and the average lifetime of the fluorophore population (the decay time constant ). Again, we found about 200 gephyrin molecules at an average spinal cord synapse, meaning that gephyrin clustering is closely regulated and that absolute molecule numbers are maintained independently of protein expression. In addition, the reconstruction of PALM-like images from the single fluorophore blinking events enabled us to estimate protein densities at synapses. With concentrations of the density of scaffold proteins exceeded that of bound inhibitory receptor complexes several-fold (e.g., at stellate cell synapses26). The relatively large population of AMPARs at excitatory synapses has been counted directly with a quantitative STORM approach.51 Here, the signals of sparse, extrasynaptic GluA2 subunits were used to estimate the number of receptor complexes at synapses in cultured hippocampal neurons. As had been seen in earlier studies49,57 the subsynaptic distribution of AMPARs was not homogenous. The receptors were typically concentrated in nanoclusters at synapses, each containing about 27 receptors. Quantitative STORM has also been employed to count absolute numbers of Bruchpilot protein (Brp) in the active zone (AZ) of Drosophila neuromuscular junctions.58 Brp plays a crucial role for presynaptic function as it binds both channels and synaptic vesicles. In this study, primary and secondary antibody titrations were carried out to determine the labeling efficiency and the conversion factor, in order to translate fluorophore detections into molecule numbers. It was calculated that 137 Brp proteins are present at an average AZ. Of these, 75% were clustered in about 15 complexes, each containing proteins.58 Together, these studies demonstrate how quantitative data in conjunction with precise subsynaptic distributions yield essential structural information that has direct relevance for synaptic function. 8.Dark Side of Multiple DetectionsMultiple detections of the same fluorophore in consecutive movie frames make it possible to reconstruct molecule trajectories and extract diffusion parameters in live sptPALM experiments.59 In fixed samples, however, multiple detections complicate single molecule counting as described earlier. In addition to successive detections, the existence of intermittent blinking must be considered in the analysis of PALM datasets to avoid major counting artifacts. Once activated, common photoconvertible proteins such as mEos2 or Dendra2 can be either irreversibly bleached or enter into long-lasting reversible dark states that are interrupted by stochastic blinking events.55,60 As a direct consequence, single fluorophores produce clusters of temporally separated detections that can be wrongly assigned as belonging to different molecules. Recurring detections need to be grouped according to their temporal and/or spatial distribution for an accurate quantification or to further improve the localization precision of the emitting fluorophores. Calibration standards with a known number of fluorophores may be considered to validate analytical strategies.52,61 A self-evident approach to account for photoblinking is to define a correct time window to join the detections of single fluorophores [Fig. 2(e)]. A systematic analysis of PALM data has shown that the estimation of fluorophore dark times () can lead to counting errors.55 Whereas a short dark time threshold will produce overcounting by assigning blinking events to separate fluorophores, long dark times cause undercounting by merging the detections from different fluorophores, particularly in dense samples. By choosing a dark time threshold that balances over- and undercounting, the absolute density of mEos2-tagged adrenergic receptors could be measured in HeLa cells.55 However, finding the correct dark time threshold is not trivial when the kinetic properties of fluorescent proteins are not well characterized. A more detailed four-state kinetic model was used to compare the photoblinking of Dendra2 and mEos2, and to establish a photoactivation protocol that maintains a constant number of active fluorophores with optimal temporal separation.60 The results indicate that Dendra2 is a better photoconvertible probe for counting purposes as it bleaches more readily and blinks less than mEos2. The temporal distribution of blinking events was further characterized using independent time traces of purified mEos2 fluorophores.62 Based on the same four-state model, it was observed that the stochastic blinking of mEos2 followed a geometric distribution with a probability that the fluorophore is bleached. This means that the number of fluorophores within a complex is convoluted with the distribution of blinking events. Since mEos2 particles behave similarly under identical imaging conditions, the histogram of blinks produced by more than one fluorophore could be fitted with a negative binomial distribution for a known value to calculate the underlying number of molecules. The analysis of protein complexes with known stoichiometry was used to estimate the photoactivation efficiency of mEos2 (), and to identify unknown stoichiometries of membrane proteins in HeLa cells.62 One advantage of this analytical approach lies in the fact that the fitting of blinking frequencies to extract and is based on a wide range of data points and is hence more accurate than the binomial fitting of the reduced molecule counts discussed earlier. Alternatively, various mathematical algorithms (e.g., Ripley’s function and DBSCAN) have been used to analyze the spatial distribution of fluorophore detections (discussed in Ref. 53). Among these, pair correlation (PC) is a statistical approach to identify nonrandom patterns in pointillist images, be it bursts of detections of the same fluorophore or accumulations of several proteins.63 By comparing the characteristic spatial signature of multiple detections from a single fluorophore to that of genuine protein clusters, it is possible to calculate the size, density, and absolute number of molecules in protein complexes [Fig. 2(h)]. An independent evaluation of the photoactivation efficiency is required to estimate absolute molecule numbers, since PC-PALM on its own does not correct for missed photoconversions. Another algorithm, SR-Tesseler, was recently proposed to identify spatially defined clusters of detections in an unbiased fashion.64 It is based on Voronoï tessellation that segments space into polygons centered on a point or seed. If applied to PALM data, each localization acts as a seed, creating polygons with a set of parameters (area and density). Adjacent polygons can be merged based on statistical criteria, hence defining discrete objects in the pointillist image such as clusters of detections belonging to single fluorophores or groups of fluorophores. SR-Tesseler was tested on SMLM data from GlyR complexes. Sparse clusters of detections were automatically selected by applying a density threshold. The analysis suggested a GlyR stoichiometry of in agreement with the earlier interpretation of the data.54 Similarly, SR-Tesseler was used to identify AMPAR nanodomains at excitatory PSDs. The mean diameter () and number of the nanodomains per spine (2.5) was in agreement with previous analyses.51 Furthermore, spatiotemporal grouping of detections suggested that nanodomains contain on average 14 AMPARs, validating tessellation techniques for the identification of clusters of detections and groups of molecules. The same approach could be applied to measure other spatially regulated molecular parameters, such as diffusion properties of membrane receptors or the strength of receptor-scaffold interactions.65,66 The combination of super-resolution techniques with fluorescence correlation spectroscopy (FCS) is particularly promising as it joins the high spatial and the high temporal resolution of the two methods.67 FCS thus provides another analytical framework to determine not only molecule concentrations but also biophysical properties within defined subcellular compartments.68 These parameters are equally important to establish accurate models of diffusion-reaction processes at synapses. 9.ConclusionThe complex molecular events during synaptic transmission need to be described by models that account for the numbers of interacting molecules and their precise distribution, and that reflect the stochasticity of synaptic processes.1 The ultimate aim must be to gather absolute numbers of several molecule types in real time and at high spatial resolution. Among various quantitative approaches, only SMLM fulfills all these requirements. This is illustrated by recent SMLM studies that have implemented quantitative, multicolor, synapse-specific, and dynamic super-resolution imaging. In addition to single molecule counting, tracking techniques can be used to extract other pertinent quantitative parameters such as diffusion properties and binding energies.69 It is therefore anticipated that SMLM will drive an impending progress in quantitative neurobiology. AcknowledgmentsA.P. is funded by a Marie-Curie ITN network grant (NPlast). We thank Nadine Schibille and Antoine Triller for critical reading of the manuscript. We would like to apologize to those researchers whose contributions to the discussed topics could not be considered. ReferencesC. Ribrault, K. Sekimoto and A. Triller,
“From the stochasticity of molecular processes to the variability of synaptic transmission,”
Nat. Rev. Neurosci., 12
(7), 375
–387
(2011). http://dx.doi.org/10.1038/nrn3025 Google Scholar
S. Okabe,
“Molecular anatomy of the postsynaptic density,”
Mol. Cell. Neurosci., 34
(4), 503
–518
(2007). http://dx.doi.org/10.1016/j.mcn.2007.01.006 Google Scholar
A. Fisher-Lavie and N. E. Ziv,
“Matching dynamics of presynaptic and postsynaptic scaffolds,”
J. Neurosci., 33
(32), 13094
–13100
(2013). http://dx.doi.org/10.1523/JNEUROSCI.2144-13.2013 Google Scholar
J. M. Levy et al.,
“Synaptic consolidation normalizes AMPAR quantal size following MAGUK loss,”
Neuron, 87
(3), 534
–548
(2015). http://dx.doi.org/10.1016/j.neuron.2015.07.015 NERNET 0896-6273 Google Scholar
P. Paoletti, C. Bellone and Q. Zhou,
“NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease,”
Nat. Rev. Neurosci., 14
(6), 383
–400
(2013). http://dx.doi.org/10.1038/nrn3504 Google Scholar
H. Monyer et al.,
“Heteromeric NMDA receptors: molecular and functional distinction of subtypes,”
Science, 256
(5060), 1217
–1221
(1992). http://dx.doi.org/10.1126/science.256.5060.1217 SCIEAS 0036-8075 Google Scholar
T. J. Hausrat et al.,
“Radixin regulates synaptic GABAA receptor density and is essential for reversal learning and short-term memory,”
Nat. Commun., 6 6872
(2015). http://dx.doi.org/10.1038/ncomms7872 Google Scholar
V. B. Caraiscos et al.,
“Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors,”
Proc. Natl. Acad. Sci. U. S. A., 101
(10), 3662
–3667
(2004). http://dx.doi.org/10.1073/pnas.0307231101 Google Scholar
V. Tretter et al.,
“Gephyrin, the enigmatic organizer at GABAergic synapses,”
Front Cell Neurosci., 6 23
(2012). http://dx.doi.org/10.3389/fncel.2012.00023 Google Scholar
F. Pfeiffer, D. Graham and H. Betz,
“Purification by affinity chromatography of the glycine receptor of rat spinal cord,”
J. Biol. Chem., 257
(16), 9389
–9393
(1982). Google Scholar
H. Betz et al.,
“How to build a glycinergic postsynaptic membrane,”
J. Cell Sci. Suppl., 15 23
–25
(1991). http://dx.doi.org/10.1242/jcs.1991.Supplement_15.4 Google Scholar
D. Langosch, L. Thomas and H. Betz,
“Conserved quaternary structure of ligand-gated ion channels: the postsynaptic glycine receptor is a pentamer,”
Proc. Natl. Acad. Sci. U. S. A., 85
(19), 7394
–7398
(1988). http://dx.doi.org/10.1073/pnas.85.19.7394 Google Scholar
P. J. Corringer et al.,
“Structure and pharmacology of pentameric receptor channels: from bacteria to brain,”
Structure, 20
(6), 941
–956
(2012). http://dx.doi.org/10.1016/j.str.2012.05.003 STRUE6 0969-2126 Google Scholar
G. E. Craft, A. Chen and A. C. Nairn,
“Recent advances in quantitative neuroproteomics,”
Methods, 61
(3), 186
–218
(2013). http://dx.doi.org/10.1016/j.ymeth.2013.04.008 MTHDE9 1046-2023 Google Scholar
J. C. Trinidad et al.,
“Activity-dependent protein dynamics define interconnected cores of co-regulated postsynaptic proteins,”
Mol. Cell. Proteomics, 12
(1), 29
–41
(2013). http://dx.doi.org/10.1074/mcp.M112.019976 Google Scholar
D. Cheng et al.,
“Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum,”
Mol. Cell. Proteomics, 5
(6), 1158
–1170
(2006). http://dx.doi.org/10.1074/mcp.D500009-MCP200 Google Scholar
J. Schwenk et al.,
“Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain,”
Neuron, 84
(1), 41
–54
(2014). http://dx.doi.org/10.1016/j.neuron.2014.08.044 NERNET 0896-6273 Google Scholar
X. Chen et al.,
“Mass of the postsynaptic density and enumeration of three key molecules,”
Proc. Natl. Acad. Sci. U. S. A., 102
(32), 11551
–11556
(2005). http://dx.doi.org/10.1073/pnas.0505359102 Google Scholar
J. C. Trinidad et al.,
“Quantitative analysis of synaptic phosphorylation and protein expression,”
Mol. Cell. Proteomics, 7
(4), 684
–696
(2008). http://dx.doi.org/10.1074/mcp.M700170-MCP200 Google Scholar
E. Neher and B. Sakmann,
“Single-channel currents recorded from membrane of denervated frog muscle fibres,”
Nature, 260
(5554), 799
–802
(1976). http://dx.doi.org/10.1038/260799a0 Google Scholar
O. P. Hamill et al.,
“Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches,”
Pflugers Arch., 391
(2), 85
–100
(1981). http://dx.doi.org/10.1007/BF00656997 Google Scholar
F. A. Edwards, A. Konnerth and B. Sakmann,
“Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch-clamp study,”
J. Physiol., 430 213
–249
(1990). http://dx.doi.org/10.1113/jphysiol.1990.sp018289 Google Scholar
F. J. Sigworth,
“The variance of sodium current fluctuations at the node of Ranvier,”
J. Physiol., 307 97
–129
(1980). http://dx.doi.org/10.1113/jphysiol.1980.sp013426 Google Scholar
P. Jonas, G. Major and B. Sakmann,
“Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus,”
J. Physiol., 472 615
–663
(1993). http://dx.doi.org/10.1113/jphysiol.1993.sp019965 Google Scholar
J. H. Singer and A. J. Berger,
“Contribution of single-channel properties to the time course and amplitude variance of quantal glycine currents recorded in rat motoneurons,”
J. Neurophysiol., 81
(4), 1608
–1616
(1999). Google Scholar
Z. Nusser, S. Cull-Candy and M. Farrant,
“Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude,”
Neuron, 19
(3), 697
–709
(1997). http://dx.doi.org/10.1016/S0896-6273(00)80382-7 NERNET 0896-6273 Google Scholar
Z. Nusser et al.,
“Relative densities of synaptic and extrasynaptic GABAA receptors on cerebellar granule cells as determined by a quantitative immunogold method,”
J. Neurosci., 15
(4), 2948
–2960
(1995). Google Scholar
C. S. Chiu et al.,
“Single-molecule measurements calibrate green fluorescent protein surface densities on transparent beads for use with ‘knock-in’ animals and other expression systems,”
J. Neurosci. Methods, 105
(1), 55
–63
(2001). http://dx.doi.org/10.1016/S0165-0270(00)00354-X Google Scholar
C. S. Chiu et al.,
“Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1-green fluorescent protein fusions,”
J. Neurosci., 22
(23), 10251
–10266
(2002). Google Scholar
Y. Sugiyama et al.,
“Determination of absolute protein numbers in single synapses by a GFP-based calibration technique,”
Nat. Methods, 2
(9), 677
–684
(2005). http://dx.doi.org/10.1038/nmeth783 Google Scholar
T. Kuriu et al.,
“Differential control of postsynaptic density scaffolds via actin-dependent and -independent mechanisms,”
J. Neurosci., 26
(29), 7693
–7706
(2006). http://dx.doi.org/10.1523/JNEUROSCI.0522-06.2006 Google Scholar
C. G. Specht et al.,
“Regulation of glycine receptor diffusion properties and gephyrin interactions by protein kinase C,”
EMBO J., 30
(18), 3842
–3853
(2011). http://dx.doi.org/10.1038/emboj.2011.276 EMJODG 0261-4189 Google Scholar
M. H. Ulbrich and E. Y. Isacoff,
“Subunit counting in membrane-bound proteins,”
Nat. Methods, 4
(4), 319
–321
(2007). http://dx.doi.org/10.1038/nmeth1024 Google Scholar
B. Laube, J. Kuhse and H. Betz,
“Evidence for a tetrameric structure of recombinant NMDA receptors,”
J. Neurosci., 18
(8), 2954
–2961
(1998). Google Scholar
C. H. Lee et al.,
“NMDA receptor structures reveal subunit arrangement and pore architecture,”
Nature, 511
(7508), 191
–197
(2014). http://dx.doi.org/10.1038/nature13548 Google Scholar
E. Karakas and H. Furukawa,
“Crystal structure of a heterotetrameric NMDA receptor ion channel,”
Science, 344
(6187), 992
–997
(2014). http://dx.doi.org/10.1126/science.1251915 SCIEAS 0036-8075 Google Scholar
P. D. Simonson et al.,
“Counting bungarotoxin binding sites of nicotinic acetylcholine receptors in mammalian cells with high signal/noise ratios,”
Biophys. J., 99
(10), L81
–L83
(2010). http://dx.doi.org/10.1016/j.bpj.2010.08.076 Google Scholar
P. Hastie et al.,
“AMPA receptor/TARP stoichiometry visualized by single-molecule subunit counting,”
Proc. Natl. Acad. Sci. U. S. A., 110
(13), 5163
–5168
(2013). http://dx.doi.org/10.1073/pnas.1218765110 Google Scholar
J. Schwenk et al.,
“High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes,”
Neuron, 74
(4), 621
–633
(2012). http://dx.doi.org/10.1016/j.neuron.2012.03.034 NERNET 0896-6273 Google Scholar
N. Durisic et al.,
“Stoichiometry of the human glycine receptor revealed by direct subunit counting,”
J. Neurosci., 32
(37), 12915
–12920
(2012). http://dx.doi.org/10.1523/JNEUROSCI.2050-12.2012 Google Scholar
J. Kuhse et al.,
“Assembly of the inhibitory glycine receptor: identification of amino acid sequence motifs governing subunit stoichiometry,”
Neuron, 11
(6), 1049
–1056
(1993). http://dx.doi.org/10.1016/0896-6273(93)90218-G NERNET 0896-6273 Google Scholar
V. Burzomato et al.,
“Stoichiometry of recombinant heteromeric glycine receptors revealed by a pore-lining region point mutation,”
Recept. Channels, 9
(6), 353
–361
(2003). http://dx.doi.org/10.3109/714041016 Google Scholar
J. Grudzinska et al.,
“The beta subunit determines the ligand binding properties of synaptic glycine receptors,”
Neuron, 45
(5), 727
–739
(2005). http://dx.doi.org/10.1016/j.neuron.2005.01.028 NERNET 0896-6273 Google Scholar
Z. Yang et al.,
“Stoichiometry and subunit arrangement of alpha1beta glycine receptors as determined by atomic force microscopy,”
Biochemistry, 51
(26), 5229
–5231
(2012). http://dx.doi.org/10.1021/bi300063m Google Scholar
J. Tonnesen and U. V. Nagerl,
“Superresolution imaging for neuroscience,”
Exp. Neurol., 242 33
–40
(2013). http://dx.doi.org/10.1016/j.expneurol.2012.10.004 Google Scholar
E. Betzig et al.,
“Imaging intracellular fluorescent proteins at nanometer resolution,”
Science, 313
(5793), 1642
–1645
(2006). http://dx.doi.org/10.1126/science.1127344 SCIEAS 0036-8075 Google Scholar
S. T. Hess, T. P. Girirajan and M. D. Mason,
“Ultra-high resolution imaging by fluorescence photoactivation localization microscopy,”
Biophys. J., 91
(11), 4258
–4272
(2006). http://dx.doi.org/10.1529/biophysj.106.091116 Google Scholar
M. J. Rust, M. Bates and X. Zhuang,
“Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM),”
Nat. Methods, 3
(10), 793
–795
(2006). http://dx.doi.org/10.1038/nmeth929 Google Scholar
A. Dani et al.,
“Superresolution imaging of chemical synapses in the brain,”
Neuron, 68
(5), 843
–856
(2010). http://dx.doi.org/10.1016/j.neuron.2010.11.021 NERNET 0896-6273 Google Scholar
C. G. Specht, I. Izeddin, M. Dahan,
“Visualizing the ultrastructures and dynamics of synapses by single-molecule nanoscopy,”
Nanoscale Imaging of Synapses: New Concepts and Opportunities, 84 75
–91 Springer, New York
(2014). Google Scholar
D. Nair et al.,
“Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95,”
J. Neurosci., 33
(32), 13204
–13224
(2013). http://dx.doi.org/10.1523/JNEUROSCI.2381-12.2013 Google Scholar
M. Renz et al.,
“Plasticity of the asialoglycoprotein receptor deciphered by ensemble FRET imaging and single-molecule counting PALM imaging,”
Proc. Natl. Acad. Sci. U. S. A., 109
(44), E2989
–E2997
(2012). http://dx.doi.org/10.1073/pnas.1211753109 Google Scholar
H. Deschout et al.,
“Progress in quantitative single-molecule localization microscopy,”
Histochem. Cell Biol., 142
(1), 5
–17
(2014). http://dx.doi.org/10.1007/s00418-014-1217-y Google Scholar
N. Durisic et al.,
“Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate,”
Nat. Methods, 11
(2), 156
–162
(2014). http://dx.doi.org/10.1038/nmeth.2784 Google Scholar
P. Annibale et al.,
“Quantitative photo activated localization microscopy: unraveling the effects of photoblinking,”
PLoS One, 6
(7), e22678
(2011). http://dx.doi.org/10.1371/journal.pone.0022678 POLNCL 1932-6203 Google Scholar
C. G. Specht et al.,
“Quantitative nanoscopy of inhibitory synapses: counting gephyrin molecules and receptor binding sites,”
Neuron, 79
(2), 308
–321
(2013). http://dx.doi.org/10.1016/j.neuron.2013.05.013 NERNET 0896-6273 Google Scholar
M. Masugi-Tokita et al.,
“Number and density of AMPA receptors in individual synapses in the rat cerebellum as revealed by SDS-digested freeze-fracture replica labeling,”
J. Neurosci., 27
(8), 2135
–2144
(2007). http://dx.doi.org/10.1523/JNEUROSCI.2861-06.2007 Google Scholar
N. Ehmann et al.,
“Quantitative super-resolution imaging of Bruchpilot distinguishes active zone states,”
Nat. Commun., 5 4650
(2014). http://dx.doi.org/10.1038/ncomms5650 Google Scholar
S. Manley et al.,
“High-density mapping of single-molecule trajectories with photoactivated localization microscopy,”
Nat. Methods, 5
(2), 155
–157
(2008). http://dx.doi.org/10.1038/nmeth.1176 Google Scholar
S. H. Lee et al.,
“Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM),”
Proc. Natl. Acad. Sci. U. S. A., 109
(43), 17436
–17441
(2012). http://dx.doi.org/10.1073/pnas.1215175109 Google Scholar
K. Finan, A. Raulf and M. Heilemann,
“A set of homo-oligomeric standards allows accurate protein counting,”
Angew. Chem. Int. Ed. Engl., 54
(41), 12049
–12052
(2015). http://dx.doi.org/10.1002/anie.201505664 Google Scholar
F. Fricke et al.,
“One, two or three? Probing the stoichiometry of membrane proteins by single-molecule localization microscopy,”
Sci. Rep., 5 14072
(2015). http://dx.doi.org/10.1038/srep14072 Google Scholar
P. Sengupta et al.,
“Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis,”
Nat. Methods, 8
(11), 969
–975
(2011). http://dx.doi.org/10.1038/nmeth.1704 Google Scholar
F. Levet et al.,
“SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data,”
Nat. Methods, 12
(11), 1065
–1071
(2015). http://dx.doi.org/10.1038/nmeth.3579 Google Scholar
N. Hoze et al.,
“Heterogeneity of AMPA receptor trafficking and molecular interactions revealed by superresolution analysis of live cell imaging,”
Proc. Natl. Acad. Sci. U. S. A., 109
(42), 17052
–17057
(2012). http://dx.doi.org/10.1073/pnas.1204589109 Google Scholar
J. B. Masson et al.,
“Mapping the energy and diffusion landscapes of membrane proteins at the cell surface using high-density single-molecule imaging and Bayesian inference: application to the multiscale dynamics of glycine receptors in the neuronal membrane,”
Biophys. J., 106
(1), 74
–83
(2014). http://dx.doi.org/10.1016/j.bpj.2013.10.027 Google Scholar
A. P. Singh and T. Wohland,
“Applications of imaging fluorescence correlation spectroscopy,”
Curr. Opin. Chem. Biol., 20 29
–35
(2014). http://dx.doi.org/10.1016/j.cbpa.2014.04.006 Google Scholar
C. Eggeling et al.,
“Direct observation of the nanoscale dynamics of membrane lipids in a living cell,”
Nature, 457
(7233), 1159
–1162
(2009). http://dx.doi.org/10.1038/nature07596 Google Scholar
C. Salvatico, C. G. Specht and A. Triller,
“Synaptic receptor dynamics: from theoretical concepts to deep quantification and chemistry in cellulo,”
Neuropharmacology, 88 2
–9
(2015). http://dx.doi.org/10.1016/j.neuropharm.2014.09.020 NEPHBW 0028-3908 Google Scholar
BiographyAngela Patrizio studied medical biotechnology and neuroscience at the Universities of Udine and Trieste, Italy. She joined Antoine Triller’s group at the ENS in Paris in 2012 to pursue her doctoral studies, funded by a Marie-Curie ITN network grant (NPlast). Her current research is focused on the development of new analytical tools based on super-resolution PALM microscopy, including strategies to infer absolute numbers of synaptic components in spinal cord neurons using single-molecule localization microscopy. Christian G. Specht developed an interest in the dynamic regulation of synaptic components during his doctoral research in the group of Ralf Schoepfer at UCL. Following a postdoctoral fellowship at Stanford University, he joined Antoine Triller’s group at the ENS in Paris to conduct further research into the plasticity of inhibitory synapses using single molecule imaging strategies. In 2011, he was appointed as a research scientist at the French National Institute of Health and Medical Research (Inserm). |
|||||||||||||||||||||||||||||||||||||||||||||



