|
|
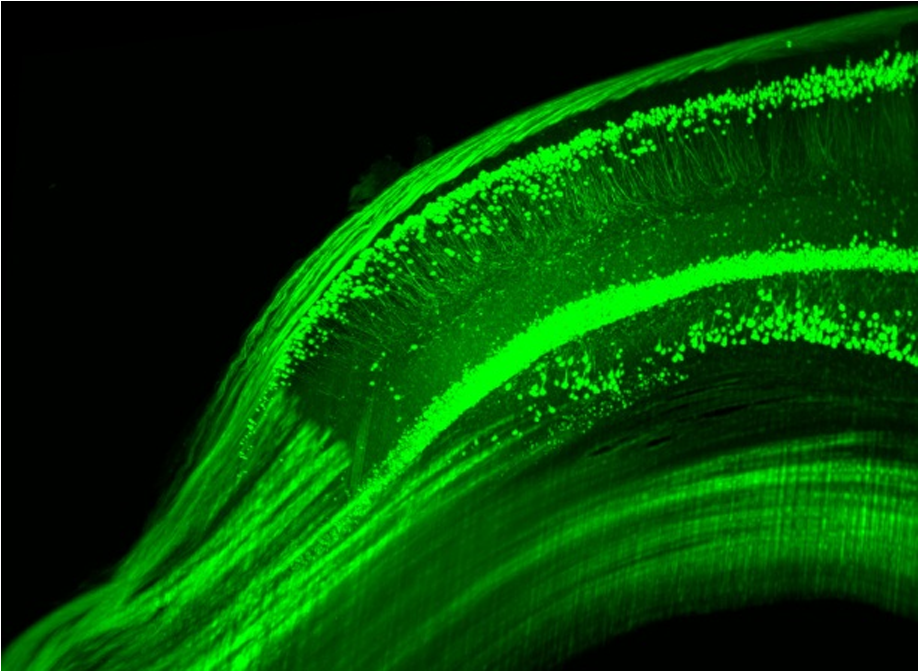
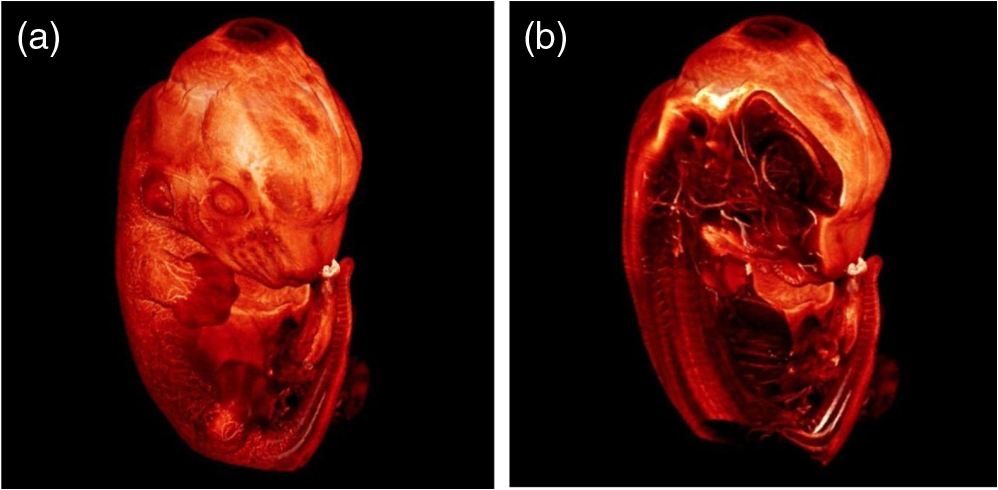
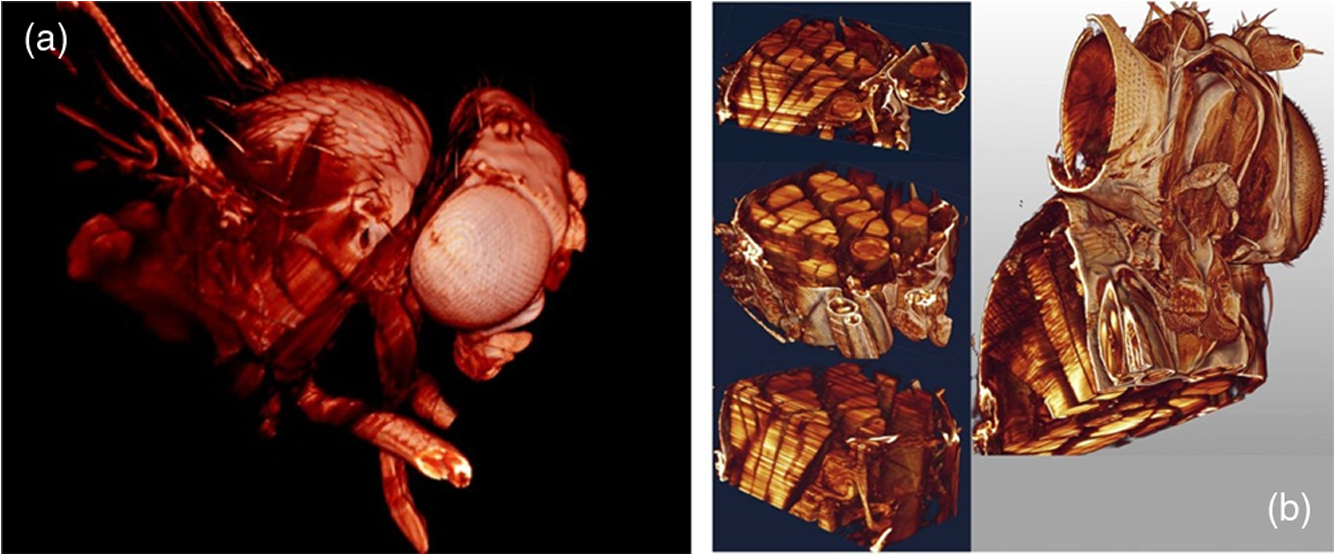
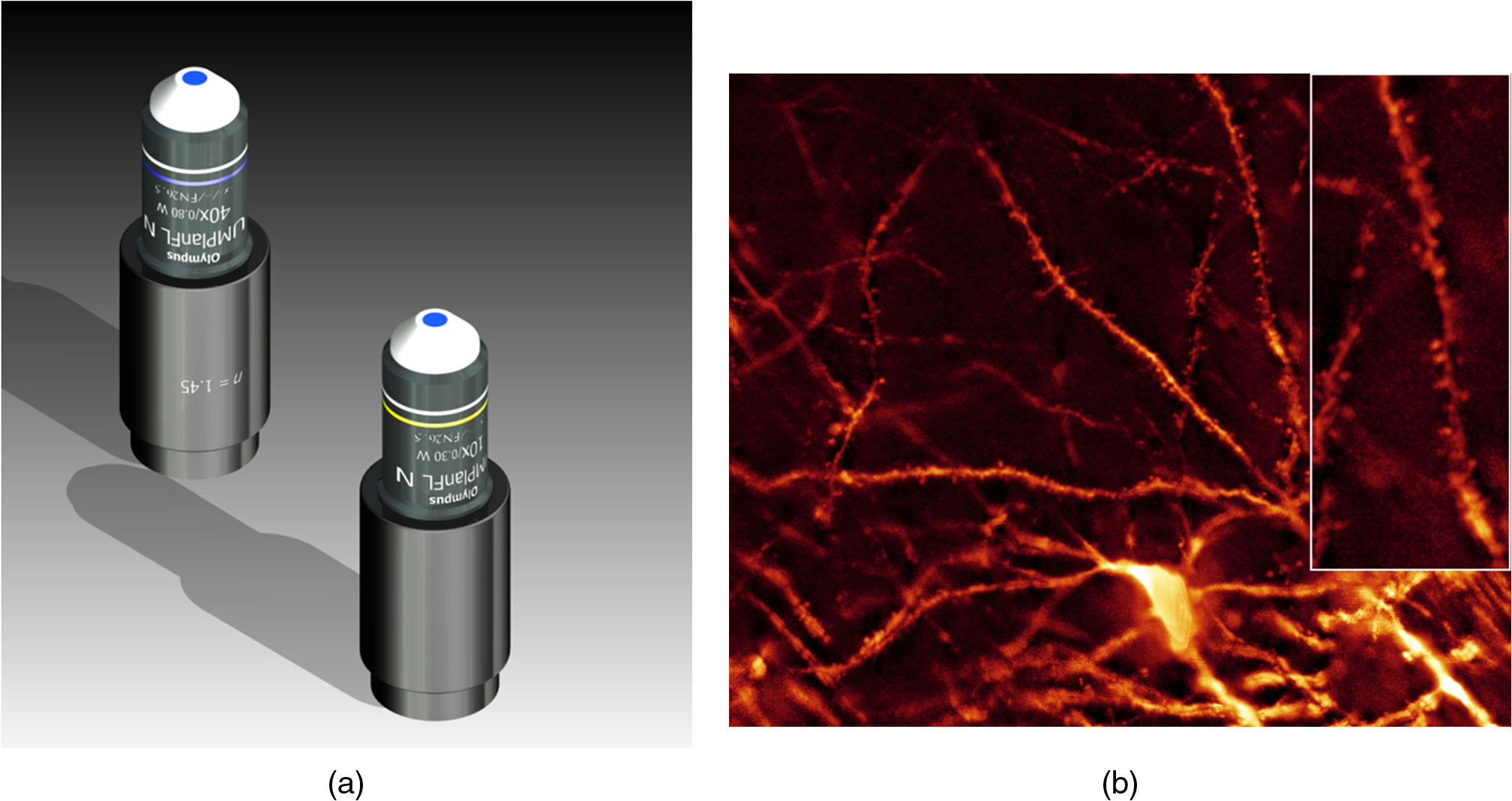
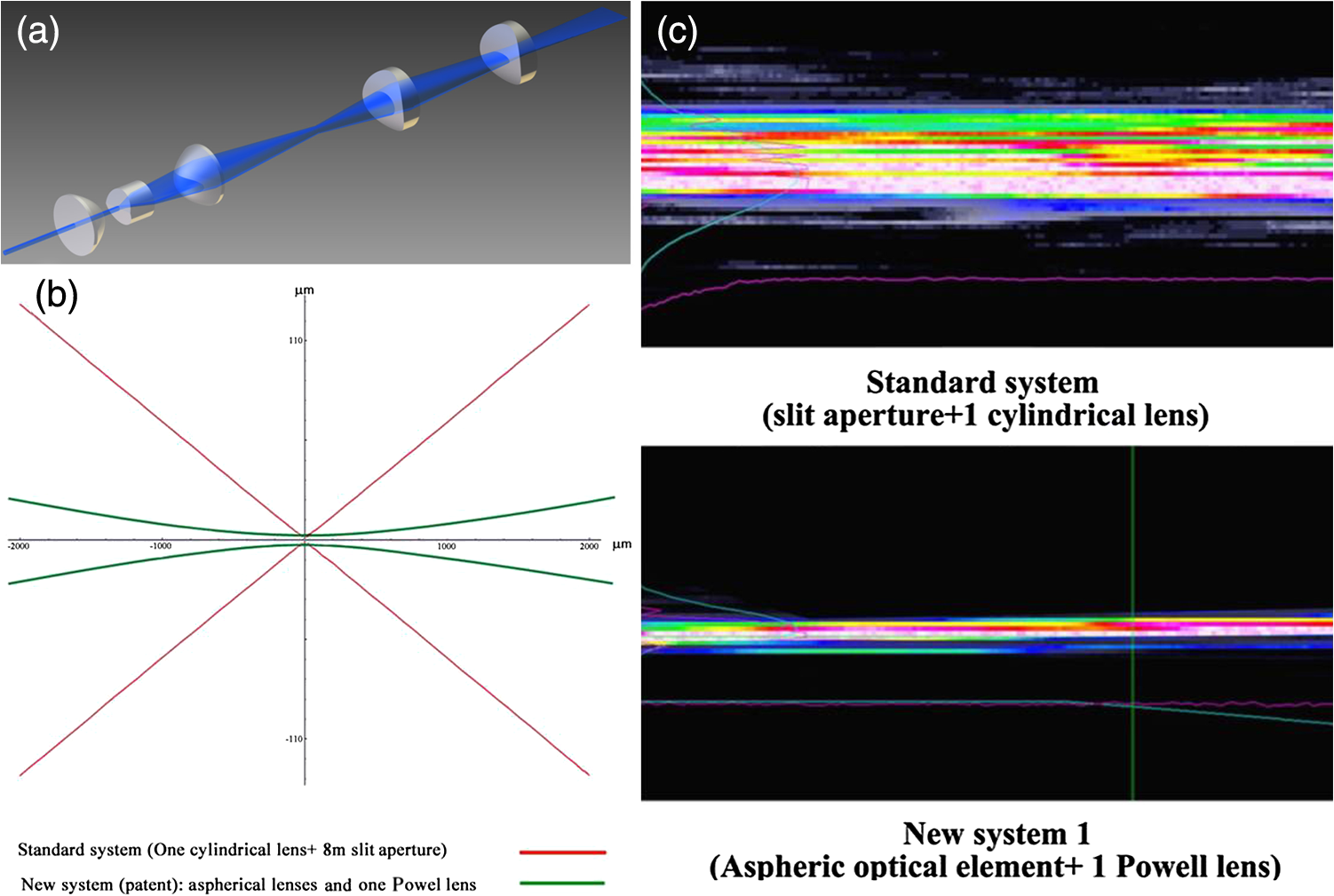
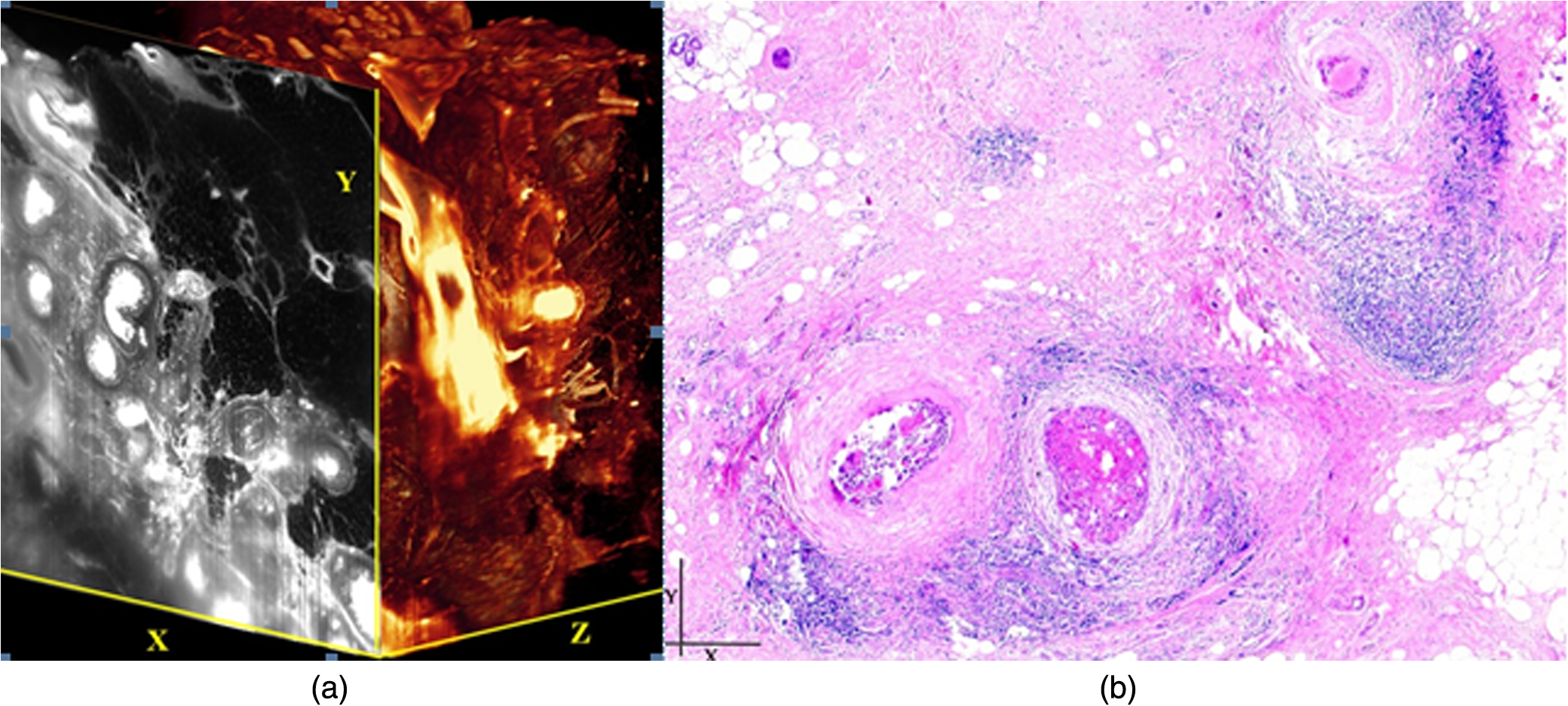
1.Background of Imaging DevelopmentIn the last decades, many new brain imaging techniques have been developed.1–4 However, whole-brain imaging [e.g., magnetic resonance imaging (MRI)] has been restricted to the macroscopic scale without single-cell resolution. This holds true for both anatomical and functional imaging. On a microscopic scale, confocal and two-photon microscopy both have penetration depths of less than 1 mm and thus cannot image large specimens such as whole brains.1 Therefore, approaches such as combination of two-photon microscopy with a milling machine were used to image whole mice hearts with cellular resolution.5 The standard procedure is, however, still histological sectioning. Unfortunately, three-dimensional (3-D) reconstructions of such sections are a rather laborious and difficult task. Also, computer-based approaches are not yet commonplace.6,7 In this context, the idea of serial sectioning has been recently revived. Ribbons of sections were collected and automatically imaged.8 Although the results of this approach are quite impressive, the method is certainly far from being routine. Another interesting approach is the use of enface imaging of resin-embedded brains containing fluorescent neurons and subsequent 3-D reconstruction (micro-optical sectioning tomography). This technique allowed brain-wide tracing of axonal projections.9–11 A very clever new approach is to use the pH dependence of green fluorescent protein (GFP) fluorescence of the neurons by manipulating the pH of the embedding resin, resulting in optical sectioning.12 However, as these techniques use massive mechanical sectioning, they may be not easy to implement. A further optical technique is optical projection tomography, but it is generally limited to a resolution of about .13 We therefore developed an alternative technique, ultramicroscopy, which allowed us to observe macroscopic specimens with microscopic resolution. Ultramicroscopy combines two 100-year-old techniques, light sheet microscopy,14 and chemical clearing of biological specimens.15 2.Development of UltramicroscopyA prominent aim of ultramicroscopy is to image neuronal networks in the whole brain with cellular resolution including long range connections. A prerequisite for this was the ability to make whole, fixed preparations transparent, and to be able to image these large specimens. Large fields of view necessitate objectives with low numerical apertures (NAs). However, confocal microscopy is difficult using objectives with NA about 0.1. In ultramicroscopy, the specimen is illuminated from the side by a thin light sheet generated by one or more cylinder lenses (Fig. 1). If this light sheet is made sufficiently thin, all parts of the specimen above or below the focal plane are in the dark and thus no out of focus light is generated.16 This straightforward approach has the further advantage that no light from out of focus planes has to be excluded further down the imaging path by a pinhole as in confocal microscopy. Hence, no superfluous light from out-of-focus planes contributes to photobleaching in the specimen. In contrast to confocal microscopy, the illumination and observation pathways are separated in ultramicroscopy. Therefore, low NA objectives can be used to provide large fields of view. To obtain transparent specimens, we applied the special clearing technique which was invented at the end of the 19th century.15 The principle of this clearing technique depends on imbuing the specimen with a medium having the same refractive index as protein.17 Thus, the refractive indices of the intra- and extracellular compartments of the specimen become equal. If the specimen becomes transparent, the light absorption significantly reduces and light can propagate through the specimen unhindered without scattering. This clearing technique enables us to make transparent complete mouse embryos, up to 2 cm in size, as well as complete mouse brains and entire adult drosophilae. 2.1.Visualization of Neurons in Hippocampus and Whole BrainUsing ultramicroscopy we have been able to visualize fine details of neurons and neuronal networks of the mouse brain. For this purpose, we used transgenic mice with GFP-labeled neurons, which display a high contrast ratio.18–20 We were able to visualize neuronal cell bodies in the hippocampus, including some of their dendrites, in the whole mouse brain. To obtain even higher resolution, we dissected hippocampi out of the whole brain and simultaneously visualized the detailed morphology of many neurons, which is not possible with standard fluorescence microscopy (Fig. 2). The complex dendritic network of pyramidal neurons became apparent in the optical sections. Using these sections, 3-D reconstructions of the dendritic networks were obtained including dendritic spines. Dendrites of granule cells could be also visualized through the overlying cortex.16 Neuronal cell bodies and the initial part of the apical dendrite can also be visualized without GFP staining. The fixation process causes some autofluorescence of the interstitial tissue, exempting neurons. Thus, by recording with a sensitive EM-CCD camera and inverting the contrast, neurons become visible to some extent.21 2.2.Visualization of Neurons in Spinal CordThe above-mentioned clearing method needs strong GFP expression and cannot be easily applied to strongly myelinated structures as the adult spinal cord. Therefore, we developed the method further to be able to investigate processes of neuronal regeneration in the injured spinal cord. Initially, we substituted alcohol for the dehydration process by tetrahydrofuran (THF), which proved to affect GFP fluorescence much less.22 We were able to trace single axons in the spinal cord and identify regenerative axons. In addition, we could label macrophages and microglia with different colors, YFP, and CFP and image all three colors in the same specimen. 2.3.Visualization of Mouse EmbryosMouse embryos are ideal objects for visualization as they can be rendered completely transparent by our clearing procedure due to their low content of absorbing or refracting material. In whole mouse embryos, we were able to visualize peripheral nerves by immunostaining (Fig. 3).23 The signal of the neurofilament antibody (anti-NF-160) was clearly distinguishable from the unspecific autofluorescence signal originating from the rest of the tissue. This allowed us to visualize the 3-D arrangement of nerves in peripheral limbs of the mouse embryo in collaboration with Edgar Kramer from the ZMNH in Hamburg. 2.4.Visualization of FliesUltramicroscopy can also be applied to the study of insects. Despite their strong pigmentation, flies can be made transparent by our clearing procedure and small anatomical details can be visualized. In Drosophila melanogaster, the entire body, including muscles and the optic lobes in the nervous system, becomes transparent. Muscle development in Drosophila is the subject of large studies. It was possible to reconstruct whole flies by imaging their autofluorescence (Fig. 4). We used this approach in cooperation with Frank Schnorrer of the MPI of Biochemistry in Munich to screen a large number of different strains of Drosophila melanogaster. In this way, we were able to identify the gene responsible for the development of the indirect flight muscles of Drosophila.24,25 3.Optics Development3.1.Correction of ObjectivesA severe problem for the imaging of cleared specimens is the lack of suitable objectives designed for high refractive index clearing solutions. The 3DISCO clearing has a refractive index of 1.56, out of the range of any commercially available objective. The resulting refractive index mismatch strongly degrades image quality.26 To solve this problem, we have adopted a new optical approach (WO 2015/010783 A1). We use air objectives with a magnification of up to and a NA of up to 0.45. We then equip them with a corrected dipping cap on the front side and with a complement optical unit on the back-side placed between the objective and the microscope. This enables us to obtain high resolution images with long working distance through clearing solution as shown in Fig. 5. Fig. 5(a) Images obtained with objective (NA, 0.28) in air and DBE without and with correction, (b) optical arrangement for objective (without and with correction optics), (c) objective with correction optics, and (d) three corrected air objectives, (NA, 0.14), (NA, 0.28) and (NA, 0.45). 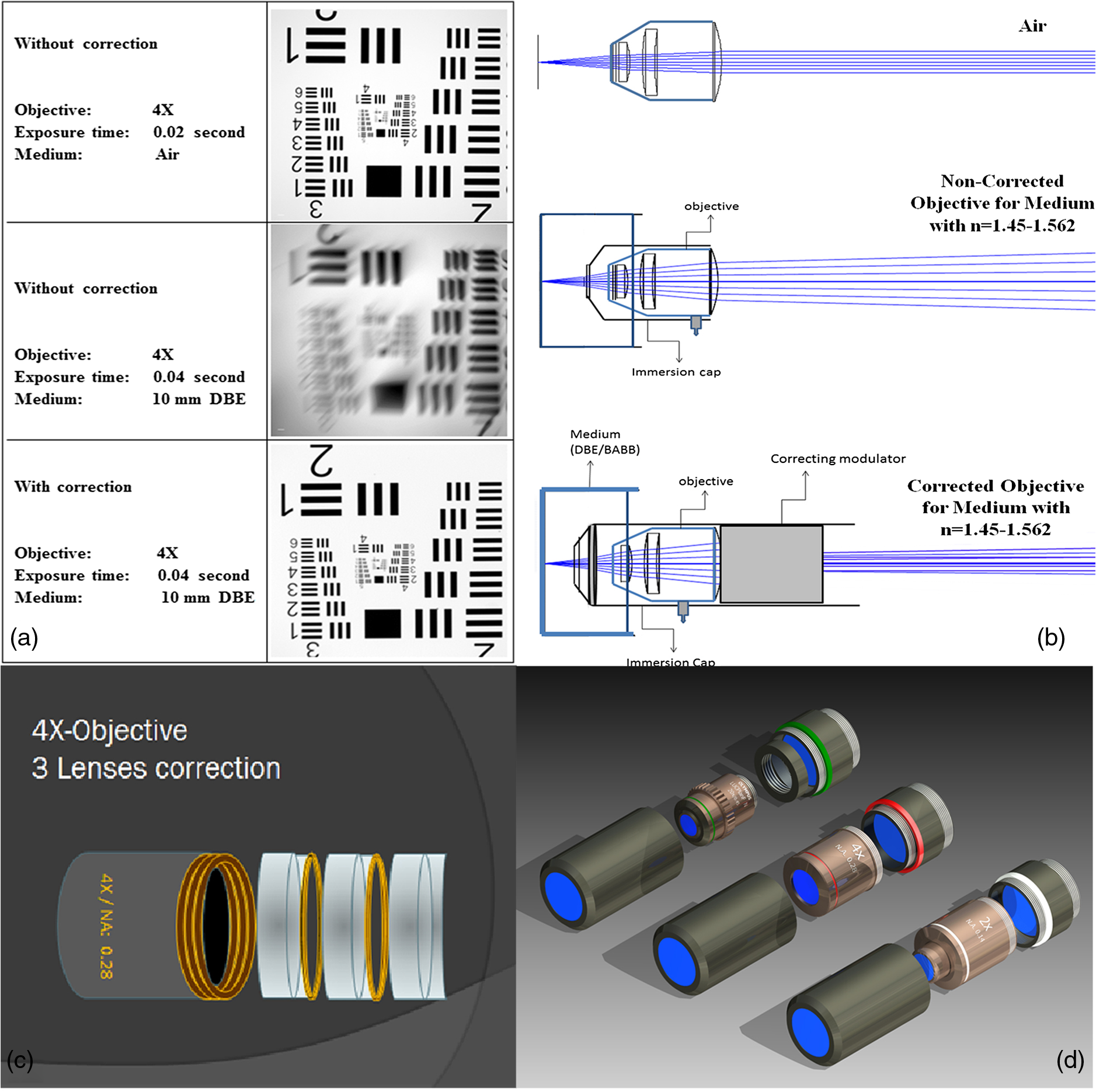 We have assembled such corrected objectives also for CLARITY clearing.27 Figure 6 shows in detail the typical neuroanatomy of the hippocampus imaged inside the whole mouse brain. Fig. 6(a) Anatomy of the hippocampus (20 optical sections) imaged in the whole mouse brain with a corrected objective (NA, 0.28). (b) Neurons in the neocortex imaged with corrected objective (300 optical sections). 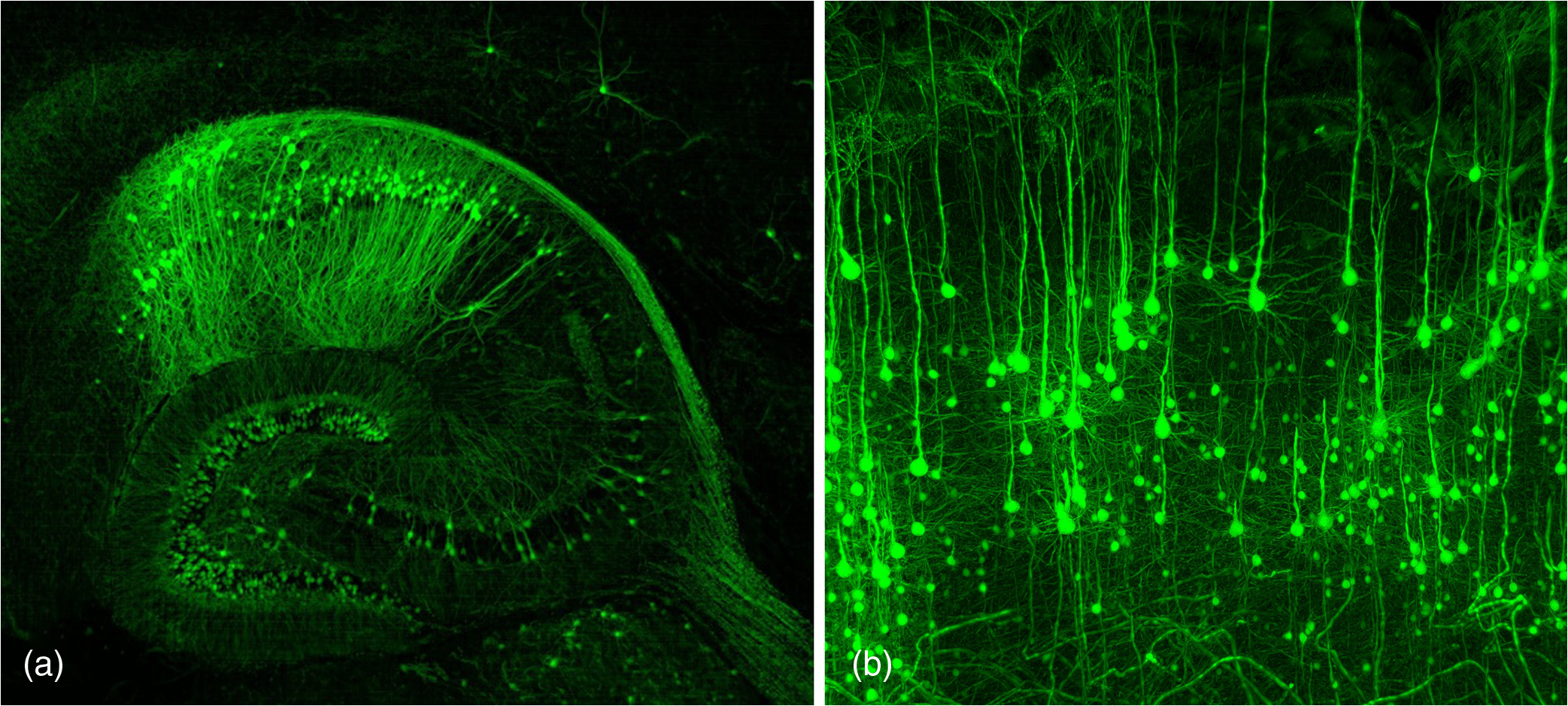 Although specialized long working distance objectives for the CLARITY clearing are becoming commercially available (Olympus); they are extremely expensive (in the 30,000 Euro range). We have thus begun to apply our postcorrection approach also to standard high-NA water-immersion objectives (e.g., , NA 0.8). They provide 3-mm working distance, which is sufficient for many biological experiments and come at a tenth of the cost of the specialized clarity objectives (Fig. 7). As these objectives are often already in the lab for patch clamping purposes, the addition of a postcorrection device may be financially a very attractive way to generate a high-NA-clarity objective. Of course, whole field and color correction may not be perfect, but this is not the case even for the expensive specialized objectives. We intend to make the described corrected air and water immersion objectives commercially available. 3.2.Generation of a Super-Thin Light SheetThe lateral resolution that is obtained in light sheet microscopy mainly depends on the NA of the microscope objectives that can be used. However, the axial resolution depends on the light sheet thickness, as the light sheet can be made thinner than the axial extension of the point-spread function of the objectives. A light sheet generated with just an ordinary cylinder lens attains a thickness considerably larger than at its ends if it is long enough to section the whole mouse brain. We have, therefore, developed a much more elaborate optical system (DE 10 2010 046 133) that uses a Powell lens and several aspheric lenses to generate a very thin, long light sheet Fig. 8.28 3.3.Further Improvement of Tissue Clearing and Preservation of Green Fluorescent Protein FluorescenceApart from optics, the clearing procedure is of paramount importance. A crucial problem related to our first clearing agent, benzyl alcohol and benzyl benzoate (BABB), is that it is not fully compatible with GFP.23 It is known that benzyl benzoate, as well as benzyl alcohol, significantly diminishes the fluorescence of GFP and, under prolonged incubation, can completely eliminate it. We, therefore, used mice with strong neuronal GFP expression and always kept the clearing times to a minimum. Because lipophilic organic clearing agents are not mixable with water, their application requires a careful dehydration of the tissue. Generally, this is done by using a graded series of ethanol. Ethanol can, however, destroy the fluorescence of GFP if the incubation times are not kept short. An optimal dehydration medium for ultramicroscopy should be mixable with water in any proportion and should not interact with GFP. We found THF to be the most promising. As a clearing agent, we found dibenzyl ether (DBE) to work best (3DISCO).29,30 BABB, DBE, and other clearing and dehydration agents tend to build organic peroxides under prolonged storage, especially in the presence of oxygen or when exposed to light. We found concentrations of peroxides of about in commercial THF and DBE. These peroxide concentrations clearly diminish the success of chemical clearing since they quench the fluorescence of GFP.31 Also, benzyl alcohol and benzyl benzoate used for the preparation of BABB are already often contaminated with peroxides when delivered by the manufacturer. Thus, it is necessary to eliminate peroxides by means of solvent extraction or column chromatography and to then store the chemicals under argon to prevent them from reacting with oxygen. The content of peroxides can easily be determined by test strips, e.g., Quantofix 25 peroxide test, Sigma-Aldrich, Germany. Finally, we began to embed our specimens in a special resin mixture in order to store them for long time and prevent bleaching. We screened various epoxy, polyester, and acrylic resins as well as different curing agents for their use as an embedding medium for cleared GFP-expressing biological tissues. Finally, we developed a transparent solid–resin formulation, which maintains the specimens’ transparency and provides a constant signal-to-noise ratio even after hours of continuous laser irradiation after an initial small drop. This may be due to preserving the fluorescent structure of GFP in the resin. The refractive index of the cured resin matches the refractive index of DBE () but can also easily be adapted to other clearing media. Thus, light can pass from the clearing medium to the resin and the resin-embedded specimen without refraction. The pronounced fluorescence-stabilizing effect of our resin embedding allows high-power illumination or long exposure times with virtually no loss in signal quality. Also, long-term storage of cleared GFP-expressing samples becomes possible for archiving purposes or repetitive investigations.32 4.Outlook4.1.Three-Dimensional-Superresolution Imaging with Stimulated Emission Depletion Light SheetsIt has been shown that it is possible to reduce light sheet thickness by applying stimulated emission depletion (STED) to light sheet microscopy (Friedrich et al.33). The reduction in light sheet thickness described was marginal, but also patent applications describing this approach can be found (e.g., DE 10 2009 008 646). By stepping the STED light sheet in the three planes x, y, and z, a 3-D superresolution reconstruction of the specimen should be obtainable.34 For this approach, it is pivotal that the specimen remains absolutely rigid so that it can be rotated without minute deformations. This is the case for 3DISCO cleared specimens, which become hard as epoxy resin. As the STED technology tends to bleach preparations, the antibleaching effect of the resin described above should be very helpful. The resin would further support the mechanical stability. At the end, the combination of all these technologies may allow for the reconstruction of the whole mouse brain with single synapse resolution. Data handling would then, of course, become an important isssue. 4.2.Biology: Towards Visualization of MemoryA very exciting application of ultramicroscopy would be the whole-brain visualization of the neuronal populations, which are involved in development, maintenance, and expression of fear memories in the mouse brain. Although current in vivo imaging techniques (e.g., fMRI) have been helpful in determining the brain areas responsible for particular functions, these methods are technically limited. None of them attains cellular resolution. Cellular resolution can, in principle, be achieved by visualization of immediate early gene expression. For this purpose, transgenic mice could be used, which express the fluorescent proteins eGFP or dtTomato controlled by the promoter of the activity-dependent gene cFos. It has been shown that cFos-eGFP expression could be induced in restricted brain regions after specific activation of neuronal ensembles.35,36 For this analysis, a combination of two-photon microscopy and histological sectioning was used. This combination proved to be reliable and powerful but also rather costly and limited in its resolution is practice. To allow visualization of activated neurons in the whole brain, high-resolution ultramicroscopy of cleared brains seems to be the method of choice. Using cFos–eGFP, it should be possible to reconstruct the distribution of activated neurons during fear conditioning and the recall of recent and remote fear memories. The establishment of this technique would open completely new avenues for the study of activity changes in complex biological systems. If once STED light sheet microscopy could be combined with the visualization of functional fluorescent proteins, one might dream of identifying synapses that are active during the storage of certain engrams in memory formation. 4.3.Clinical Applications of UltramicroscopyUltramicroscopy should be also applicable for diagnosis in pathology regarding cancer staging. It allows a 3-D reconstruction of malignant tissue removed during operation. Of special interest for pathologists is the nature of the tissue at the margins of the operation material. We found that in fixed tumor tissue, the structures become visible just by their autofluorescence. This is probably induced by the formalin fixation (Fig. 9). Exact identification of tumor and normal tissue will need further correlative studies with standard histological staining. However, in Fig. 9, the strong autofluorescent material in milk-duct-like structures is already prominent. As the histological stain of another part of the tumor shows, this bright material seems to be cancerous cells in the milk ducts, typical for this ductal carcinoma (DCIS) in situ. With ultramicroscopy, now the 3-D arrangement of this part of the tumor can be visualized. This first example shows that in the future, ultramicroscopy may become a valuable instrument for tumor staging in clinical medicine. AcknowledgmentsWe thank the Austrian science fund (FWF-P23102-N22) and the Hertie Stiftung (Project No. 1.02.1/04/004) for funding support. ReferencesF. Helmchen and W. Denk,
“Deep tissue two-photon microscopy,”
Nat. Methods, 2 932
–940
(2005). http://dx.doi.org/10.1038/nmeth818 1548-7091 Google Scholar
G. A. Johnson et al.,
“Morphological phenotyping with MR microscopy: the visible mouse,”
Radiology, 222 789
–793
(2002). http://dx.doi.org/10.1148/radiol.2223010531 RADLAX 0033-8419 Google Scholar
W. A. Kalender,
“CT: the unexpected evolution of an imaging modality,”
Eur Radiol. Suppl., 15
(Suppl. 4), D21
–D24
(2005). http://dx.doi.org/10.1007/s10406-005-0128-3 Google Scholar
J. M. Tyszka, S. E. Fraser and R. E. Jacobs,
“Magnetic resonance microscopy: recent advances and applications,”
Curr. Opin. Biotechnol., 16 93
–99
(2005). http://dx.doi.org/10.1016/j.copbio.2004.11.004 CUOBE3 0958-1669 Google Scholar
T. Ragan et al.,
“High-resolution whole organ imaging using two-photon tissue cytometry,”
J. Biomed. Opt., 12 014015
(2007). http://dx.doi.org/10.1117/1.2435626 Google Scholar
J. Streicher et al.,
“Computer-based three-dimensional visualization of developmental gene expression,”
Nat. Genet., 25 147
–152
(2000). http://dx.doi.org/10.1038/75989 NGENEC 1061-4036 Google Scholar
W. J. Weninger and T. Mohun,
“Phenotyping transgenic embryos: a rapid 3-D screening method based on episcopic fluorescence image capturing,”
Nat. Genet., 30 59
–65
(2002). http://dx.doi.org/10.1038/ng785 NGENEC 1061-4036 Google Scholar
S. Micheva and S. J. Smith,
“Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits,”
Neuron, 55 25
–36
(2007). http://dx.doi.org/10.1016/j.neuron.2007.06.014 NERNET 0896-6273 Google Scholar
H. Gong et al.,
“Continuously tracing brain-wide long-distance axonal projections in mice at a one-micron voxel resolution,”
NeuroImage, 74 87
–98
(2013). http://dx.doi.org/10.1016/j.neuroimage.2013.02.005 NEIMEF 1053-8119 Google Scholar
A. Li et al.,
“Micro-optical sectioning tomography to obtain a high-resolution atlas of the mouse brain,”
Science, 330 404
–1408
(2010). http://dx.doi.org/10.1126/science.1191776 SCIEAS 0036-8075 Google Scholar
T. Zheng et al.,
“Visualization of brain circuits using two-photon fluorescence micro-optical sectioning tomography,”
Opt. Express, 21 9839
–9850
(2013). http://dx.doi.org/10.1364/OE.21.009839 OPEXFF 1094-4087 Google Scholar
H. Xiong et al.,
“Chemical reactivation of quenched fluorescent protein molecules enables resin-embedded fluorescence microimaging,”
Nat. Commun., 5 3992
(2013). http://dx.doi.org/10.1038/ncomms4992 NCAOBW 2041-1723 Google Scholar
J. Sharpe et al.,
“Optical projection tomography as a tool for 3D microscopy and gene expression studies,”
Science, 296 541
–545
(2002). http://dx.doi.org/10.1126/science.1068206 SCIEAS 0036-8075 Google Scholar
H. Siedentopf and R. Zsigmondy,
“Über Sichtbarmachung und Größenbestimmung ultramikroskopischer Teilchen, mit besonderer Anwendung auf Goldrubingläser,”
Ann. Phys., 10 1
–39
(1903). http://dx.doi.org/10.1002/andp.19023150102 ANPYA2 0003-3804 Google Scholar
W. Spalteholz,
“Über das Durchsichtigmachen von menschlichen und tierischen Präparaten,”
Hierzel, Leipzig, 2 91
(1914). Google Scholar
H. U. Dodt et al.,
“Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain,”
Nat. Methods, 4 331
–336
(2007). http://dx.doi.org/10.1038/nmeth1036 1548-7091 Google Scholar
J. A. Dent, A. G. Polson and M. W. Klymkowski,
“A whole-mount immunocytochemical analysis of the expression of the intermediate filament protein vimentin in Xenopus,”
Development, 105 61
–74
(1989). Google Scholar
R. N. Day and F. Schaufele,
“Fluorescent protein tools for studying protein dynamics in living cells: a review,”
J. Biomed. Opt., 13 031202
(2008). http://dx.doi.org/10.1117/1.2939093 JBOPFO 1083-3668 Google Scholar
G. Feng et al.,
“Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP,”
Neuron, 28 41
–51
(2000). http://dx.doi.org/10.1016/S0896-6273(00)00084-2 NERNET 0896-6273 Google Scholar
J. Livet et al.,
“Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system,”
Nature, 450 56
–62
(2007). http://dx.doi.org/10.1038/nature06293 Google Scholar
U. Leischner, W. Zieglgänsberger and H. U. Dodt,
“Resolution of ultramicroscopy and field of view analysis,”
PLoS One, 4 e5785
(2009). http://dx.doi.org/10.1371/journal.pone.0005785 POLNCL 1932-6203 Google Scholar
A. Ertürk et al.,
“Three-dimensional imaging of solvent-cleared organs using 3DISCO,”
Nat. Protoc., 7 1986
–1995
(2012). http://dx.doi.org/10.1038/nprot.2012.119 1754-2189 Google Scholar
K. Becker et al.,
“Ultramicroscopy: 3D reconstruction of large microscopical specimens,”
J. Biophotonics, 1
(1), 36
–42
(2008). http://dx.doi.org/10.1002/jbio.200710011 Google Scholar
N. Jährling et al.,
“Three-dimensional reconstruction and segmentation of intact drosophila by ultramicroscopy,”
Front. Syst. Neurosci., 4 1
–4
(2010). http://dx.doi.org/10.3389/neuro.06.001.2010 Google Scholar
C. Schönbauer et al.,
“Spalt mediates an evolutionarily conserved switch to fibrillar muscle fate in insects,”
Nature, 479 406
–409
(2011). http://dx.doi.org/10.1038/nature10559 Google Scholar
H. Jacobsen and S. W. Hell,
“Effect of the specimen refractive index on the imaging of a confocal fluorescence microscope employing high aperture oil immersion lenses,”
Bioimaging, 3 39
–47
(1995). http://dx.doi.org/10.1002/1361-6374(199503)3:1<39::AID-BIO5>3.3.CO;2-Z BOIMEL 0966-9051 Google Scholar
K. Chung and K. Deisserroth,
“CLARITY for mapping the nervous system,”
Nat. Methods, 10 508
–513
(2013). http://dx.doi.org/10.1038/nmeth.2481 1548-7091 Google Scholar
S. Saghafi et al.,
“3D-ultramicroscopy utilizing aspheric optics,”
J. Biophotonics, 7 117
–125
(2014). http://dx.doi.org/10.1002/jbio.201300048 Google Scholar
A. Ertürk et al.,
“Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury,”
Nat. Med., 18 166
–171
(2012). http://dx.doi.org/10.1038/nm.2600 Google Scholar
A. Ertürk et al.,
“Three-dimensional imaging of solvent-cleared organs using 3DISCO,”
Nat. Protoc., 7 1986
–1995
(2012). http://dx.doi.org/10.1038/nprot.2012.119 1754-2189 Google Scholar
A. A. Alnuami et al.,
“Oxyradical-induced GFP damage and loss of fluorescence,”
Int. J. Biol. Macromol., 43 182
–186
(2008). http://dx.doi.org/10.1016/j.ijbiomac.2008.05.002 Google Scholar
K. Becker et al.,
“Reduction of photobleaching and long term archiving of chemically cleared GFP-expressing mouse brains,”
PLoS One, 9 e114149
(2014). http://dx.doi.org/10.1371/journal.pone.0114149 POLNCL 1932-6203 Google Scholar
M. Friedrich et al.,
“STED-SPIM: stimulated emission depletion improves sheet illumination microscopy resolution,”
Biophys. J., 100 L43
–L45
(2011). http://dx.doi.org/10.1016/j.bpj.2010.12.3748 Google Scholar
H. U. Dodt,
“The superresolved brain,”
Science, 347 474
–475
(2015). http://dx.doi.org/10.1126/science.aaa5084 SCIEAS 0036-8075 Google Scholar
A. L. Barth, R. C. Gerkin and K. L. Dean,
“Alteration of neuronal firing properties after in vivo experience in a FosGFP transgenic mouse,”
J. Neurosci., 24 6466
–6475
(2004). http://dx.doi.org/10.1523/JNEUROSCI.4737-03.2004 Google Scholar
Y. Kim et al.,
“Mapping social behaviour-induced brain activation at cellular resolution in the mouse,”
Cell Reports, 10 292
–305
(2015). http://dx.doi.org/10.1016/j.celrep.2014.12.014 Google Scholar
BiographyHans-Ulrich Dodt studied medicine in Freiburg/Br. and Heidelberg and physics in Heidelberg, Germany. He worked as a research assistant at the Max-Planck-Institute of Psychiatry in Munich, Department of Neurophysiology. In 1987, he received his MSc in physics at the Max-Planck-Institute of Astronomy in Heidelberg, Germany. From 1987 to 2006, he worked as a research assistant at the Max-Planck-Institute of Psychiatry. From 2005 to 2006, he was a group leader at the Max-Planck-Institute of Psychiatry. Since January 2007, he has been a full professor of bioelectronics at the Technical University of Vienna, Austria. Saiedeh Saghafi received her BSc degree in applied physics from the University of Tehran. In 1996, she started her MSc degree in optical physics at University of Sydney in professor Colin Sheppard group. She studied Gaussian beam propagation in and beyond the paraxial approximation. In 1998, she won an overseas postgraduate award to attend the PhD program in the Physics Department at Macquarie University in Australia. In 2000, she won two research awards from the Australian Optical Society and IEEE/LEOS Graduate Fellowship. In 2001, she received her PhD in modal analysis of lasers employing unstable resonators. Her thesis was nominated for the AIP Bragg gold medal for the best PhD thesis. She held a postdoctoral position for one year in CLA—Centre for lasers and Applications. Then, she worked as an assistant professor in the Biophotonics group at IAU, working on the medical applications. She is working at the University of Technology Vienna, FKE, Department of Bioelectronics on Ultramicroscopy techniques since 2008 as postdoc and since 2013 as a university assistant. Klaus Becker studied biology at the Justus-Liebig University of Gießen. In 1998, he received his PhD in theoretical biology from the Freie University of Berlin on the topic of computer simulations of vision in insects. He worked as a stipendiary at the Laboratory of Psychophysiology and Electrophysiology of Vision, Universidade Sao Paulo, Brazil. From 2000 until 2006, he held a postdoc position at the Max-Planck-Institute of Psychiatry, Munich, Germany, in the research groups Clinical Neuropharmacology and Bioimaging. Since 2007, he has worked as a senior scientist at the Vienna University of Technology, FKE, Department of Bioelectronics and at the Center of Brain Research, Medical University, Vienna, Austria. Nina Jährling studied biology at the Carl von Ossietzky University of Oldenburg (Germany). She received her PhD in 2011 for her work on “Applications of Ultramicroscopy on neurobiology and methodological improvements” from the University of Oldenburg for studies at the Technical University Vienna. Since 2012, she continued her work in the group of Hans-Ulrich Dodt, where she is working as a university assistant at the Technical University Vienna, FKE, Department for Bioelectronics, and at the Center of Brain Research, Medical University of Vienna (Austria). Christian Hahn studied biology at the Ruprecht-Karls University of Heidelberg (Germany). He received his PhD for his work on axonal path finding in the visual system, i.e., on intracellular signaling and stabilization of the cytoskeleton. At the Leibniz Institute for Age Research in Jena (Germany), he worked as a postdoc on thyroid hormone transport during early brain development. In 2012, he joined the group of Hans-Ulrich Dodt, where he is working as a postdoc at the Vienna Technical University, FKE, Department for Bioelectronics, and at the Center of Brain Research, Medical University of Vienna, Austria. Marko Pende studied molecular biology at the University of Vienna (Austria). He received his MSc at the Center for Brain Research (CBR), Medical University of Vienna (Austria) for his work on neuromyelitis optica like lesion in experimental autoimmune encephalomyelitis (EAE) mice. In 2013, he joined the group of Hans-Ulrich Dodt, where he is working as a PhD student at the CBR and at the Vienna Technical University, FKE, Department for Bioelectronics (Austria). Martina Wanis studies biology at the University of Vienna (Austria) with the focus on neuroscience. Currently, she is working in cooperation between TU Wien and the University of Vienna on the topic of chemical clearing of Drosophila to study brain development by the use of confocal and light sheet imaging under the supervision of Thomas Hummel and Hans-Ulrich Dodt. Axel Niendorf studied medicine and literature at the Universities of Bordeaux, Kiel, and Hamburg from 1979 to 1985. From 1986 to 1995, he worked at the Institute of Pathology of the University of Hamburg. Since 1995, he has been in private practice in Hamburg with a diagnostic emphasis on mammary, cervix, and lung. His particular scientific interest lies in the morphological and molecular analysis of oncology biomarkers. |